



分享:氯离子对Pb-Ag-RE合金阳极电化学行为的影响

钟晓聪, 蒋良兴 , 吕晓军, 赖延清, 李劼, 刘业翔
, 吕晓军, 赖延清, 李劼, 刘业翔
中南大学冶金与环境学院, 长沙 410083
摘要
采用恒流极化, SEM, XRD, EIS和Tafel扫描对比研究了Pb-Ag-RE合金阳极在无Cl-和含500 mg/L Cl- H2SO4溶液中的腐蚀行为和析氧行为. 结果表明, Pb-Ag-RE阳极在含Cl-电解液中生成的氧化膜层出现“火山口”状孔洞, 合金基底上分布着大量的腐蚀坑, 呈现出明显的局部腐蚀特征. 此外, Cl-的存在会减少阳极表面氧化膜层中PbO2的含量, 抑制析氧反应中间产物的生成和吸附, 进而增加析氧反应传荷阻抗. 因此, 500 mg/L的Cl-对Pb-Ag-RE合金阳极的耐腐蚀性能和析氧活性均会造成不利影响, 工业生产中应尽量降低电解液中Cl-的浓度.
关键词:
有色金属(如Zn, Cu, Co, Ni和Mn)的湿法冶金电沉积工序普遍采用硫酸电解液, 铅基合金由于在高浓度硫酸溶液和高电流密度的工作条件下稳定性好, 被广泛用作析氧阳极[1,2]. 铅基合金阳极在服役初期可以形成一层保护性的氧化膜层. 该膜层可以大大减缓合金基底的进一步氧化腐蚀, 因此阳极具有较好的稳定性和耐腐蚀性[3]. 研究[4]表明, 铅基合金阳极的耐腐蚀性受氧化膜层的结构和成分的影响, 而氧化膜层的结构和组成从本质上又由铅基合金的微观金相结构和电解液的性质决定.
为了改善铅基阳极的性能, 可以调控其微观金相结构和控制电解液的成分. 针对调控铅基合金的微观结构, 主要有三种途径: 一是优化合金元素的种类和含量[5,6]; 二是控制合金浇铸过程的冷却制度[7]; 三是机械加工和热处理[8]. 对于控制电解液成分, 可以添加一些有益的金属离子(如Mn2+和Co2+)[1,9], 同时还要尽量减少F−等对铅基阳极电化学性能有害的离子.
Zn电解工业中, 电解液中普遍含有500 mg/L左右的Cl-. 随着矿物成分的日益复杂和工业循环溶液中Cl-的积累[10], 有些电解液Cl-浓度甚至可以达到1000 mg/L[11]. 因此, 国内外越来越重视Cl-对电解过程中铅基阳极性能的影响. Fraunhofer[12]发现Cl-会与Pb2+沉淀生成PbCl2, 在更高的电位下PbCl2会被氧化成PbO2, 而且Cl2可能与O2一起析出. Ivanov等[13]综述了Cl-对Pb及铅合金阳极性能的影响, 其中报道了当Cl-浓度为100 mg/L时, Pb-Ag阳极的腐蚀与无Cl-电解液中的腐蚀相当, 而纯Pb阳极即便在含低浓度Cl-的电解液中也会剧烈腐蚀. 500 mg/L的Cl-则可以明显加剧Pb-Ag阳极的腐蚀. Hampson等[14]发现Cl-会降低膜层中SO42-的稳定性, 降低氧化膜层的质量并抑制钝化过程, 同时Cl-可以降低析氧过电位. Liu等[10]也认为Cl-会降低膜层的保护性能, 加速膜层的溶解. Cifuents等[15]则发现, 在Cu电沉积过程中, Cl-浓度低于100 mg/L时, Cl-可以减少Pb的失重和腐蚀. Tunnicliffe等[9]也报道了Cl-可以减少Pb-Ag阳极的腐蚀, 其原因是服役过程中生成的AgCl2可以包围氧化膜层, 提高膜层的耐腐蚀性能. 因此, Cl−对铅合金阳极有利还是有害目前还无定论, 需要进一步研究.
本工作通过对比研究Pb-Ag-RE合金阳极在无Cl-和含Cl- H2SO4溶液中的腐蚀行为和析氧行为, 分别探索了Cl-对Pb-Ag-RE合金的阳极电位、氧化膜层形貌和物相、腐蚀速率和腐蚀基底形貌及析氧反应动力学参数的影响, 并讨论了Cl−对Pb-Ag-RE合金阳极电化学行为的影响机制, 为湿法冶金电沉积工业电解液中Cl-浓度的调控提供参考.
1 实验方法
1.1 实验材料
工作电极选用压延Pb-Ag-RE合金. 采用PS-6电感耦合等离子体原子发射光谱仪(ICP-AES)检测得到其化学成分为Pb-0.45%Ag-0.03%RE (质量分数). 将合金线切割成10 mm × 10 mm × 5 mm的试样. 经焊接Cu导线和义齿基托树脂密封获得工作面积为1 cm2的工作电极. 对电极采用表面积为4 cm2的Pt电极, 参比电极为Hg/Hg2SO4/饱和K2SO4 (0.64 V vs 标准氢电极电位). 如无特殊说明, 文中所有电位均参照该参比电极.
采用无Cl-和含500 mg/L Cl- 2种H2SO4溶液(160 g/L)模拟工业用电解液. 电解液由分析纯H2SO4, 分析纯HCl和去离子水配制. 电解液的温度控制在(35±1) ℃. 进行电化学测试前, 采用SiC砂纸将工作电极打磨光亮, 然后用去离子水冲洗干净备用.
1.2 测试方法
采用1470 E电化学工作站记录Pb-Ag-RE阳极在2种H2SO4溶液中恒流极化(500 A/m2, Zn电沉积工业采用的电流密度) 72 h过程中的阳极电位. 恒流极化后, 阳极表面形成氧化膜层, 分别采用MIRA 3扫描电子显微镜(SEM)和D/max 2500 X-射线衍射仪(XRD)检测阳极氧化膜层的形貌和物相. 为了研究Cl-对Pb-Ag-RE阳极腐蚀速率和腐蚀特性的影响, 采用ICP-AES检测电解液中Pb2+浓度随恒流极化时间的变化. 将极化后的阳极在沸腾的糖碱溶液(20 g/L葡萄糖+100 g/L NaOH)中浸泡5 min以溶解去除阳极表面氧化膜层, 然后用SEM观察合金阳极基底的腐蚀形貌.
为了探索Cl-对Pb-Ag-RE阳极析氧行为的影响, 在Pb-Ag-RE阳极恒流极化(500 A/m2) 72 h后立即采用电化学阻抗(EIS)和Tafel扫描研究其析氧过程动力学. EIS测试中, 偏置电位为恒流极化后期的稳定阳极电位, 交流振幅为10 mV, 频率范围为100 kHz~0.1 Hz. Tafel电位扫描范围为1.20~1.45 V, 扫描速率为0.166 mV/s.
2 实验结果与讨论
2.1 阳极电位
图1所示为Pb-Ag-RE合金阳极在2种H2SO4溶液恒流极化过程中阳极电位随时间的变化. 从图1可以看出, 电解液中Cl-的存在对Pb-Ag-RE阳极电位的变化趋势没有明显的影响. 在极化初期, 阳极电位快速降低. 这一阶段主要进行的是PbSO4层的快速生长. 随着PbSO4层变厚, 覆盖度变大, 氧化膜层的阻抗增加. 因此, 电位出现短暂的回升. 随着极化时间进一步延长, 阳极电位缓慢地降低. 这是由于膜层中部分导电性差的PbSO4向导电性较好的PbO2转变, 而且PbO2有利于析氧活性位点的生成, 减小析氧反应的极化程度. 因此, 阳极电位逐渐降低. 在含500 mg/L Cl- H2SO4溶液中, Pb-Ag-RE的阳极电位均低于无Cl-电解液中的电位. 72 h极化后, 含Cl−电解液中的阳极电位约低20 mV.
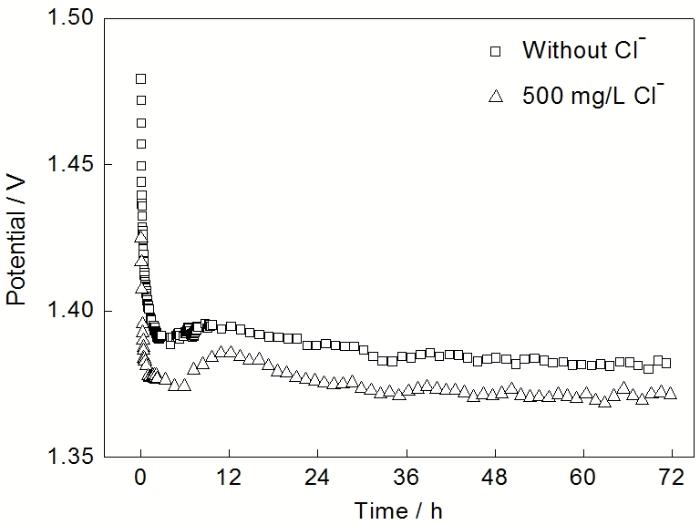
图1 Pb-Ag-RE阳极在无Cl-和含500 mg/L Cl- H2SO4溶液中恒流极化过程中阳极电位的变化
Fig.1 Anodic potential variation of Pb-Ag-RE anode during galvanostatic polarization in H2SO4 solution without Cl- and with 500 mg/L Cl-
2.2 氧化膜层
阳极氧化膜层的形貌和物相组成会影响合金基底的腐蚀速率. 这是因为膜层一方面起着物理隔离基底和电解液的作用, 膜层的孔隙率、致密度会影响电解液在膜层中的扩散传质, 进而影响基底的腐蚀. 另一方面, 按照Pavlov和Dinev[16]的理论, 氧化膜层/电解液界面上新生成的氧会从该界面向基底/膜层界面传输, 而氧空位按相反方向传输. 最终氧与Pb发生氧化反应, 而氧空位在氧化膜层/电解液界面参与电子转移. 因此, 氧化膜层的物相组成还影响新生氧在膜层中的固相传输, 进而影响基底氧化腐蚀的速率. 此外, 由于膜层/电解液界面是析氧反应进行的场所, 膜层表面的物相组成影响析氧反应的活性位点的数量和分布, 同时膜层表面积也影响异相反应的反应速率. 因此, 氧化膜层还在很大程度上决定着阳极的析氧活性. 综上所述, 有必要研究Cl−对Pb-Ag-RE合金阳极在极化过程中形成的氧化膜层的形貌和物相组成的影响.

图2 Pb-Ag-RE阳极在无Cl-和含500 mg/L Cl- H2SO4溶液中恒流极化72 h过程中生成的氧化膜层的SEM像
Fig.2 SEM images of anodic layers on Pb-Ag-RE anode obtained through 72 h galvanostatic polarization in H2SO4 solution without Cl- (a) and with 500 mg/L Cl- (b)
2.2.1 形貌 图2是Pb-Ag-RE阳极在2种电解液中恒流极化72 h后形成的氧化膜层的SEM像. 图2a呈现出典型的珊瑚礁状. 这个形貌特征主要由两方面原因导致: 一是膜层受到析氧反应生成的O2气泡的持续冲刷; 二是膜层表面的部分PbSO4和PbO2会相互转变, 由于两者摩尔体积不同, 转变过程中的膜层收缩和舒张导致膜层致密度较低. 在含Cl-电解液中, 氧化膜层呈现出不同的特征, 如图2b所示. 膜层中出现很多“火山口”状的孔洞. 而在这些孔洞周围, 膜层呈现胶状, 这与Fraunhofer[12]的报道一致. 这些区域较无Cl−电解液中获得的膜层更平整、致密. 值得注意的是, 图2a中的孔洞是由于膜层凹凸不平导致的, 这些孔洞很大部分没有贯通到膜层内部. 而对于图2b中的“火山口”状的孔洞是深入膜层内部的, 这将加剧基底的腐蚀.
2.2.2 物相 图3给出了Pb-Ag-RE阳极在2种H2SO4溶液中极化72 h后生成的氧化膜层的XRD谱. 从图3可以看出, 2种电解液中生成的氧化膜层都主要由β-PbO2和α-PbO2组成. XRD谱中未看到明显的PbSO4和非化学计量比PbOn及碱性PbSO4等物质的特征峰. 这是由于这些物相在膜层表面的含量较少或被表层PbO2所覆盖. 在含Cl−电解液中生成的氧化膜  层的a-PbO2的特征峰峰强更小, Hampson等[14]分析认为, 这是因为Cl−的存在加速了膜层的溶解, 导致膜层厚度减小. 值得注意的是, 在XRD谱中可以看到金属Pb的特征峰, 表明膜层不致密, 裸露出部分铅基合金基底. 还有一种解释是, 由于膜层厚度较薄, 导致X-射线穿透膜层, 检测到基底的信号. 含Cl−电解液中的膜层的Pb的特征峰更明显, 而且峰强更大. 这是由于Cl−的存在使膜层中有许多“火山口”状的孔洞, 降低了膜层的致密度, 使得更多的基底裸露. 此外, Cl−也可能减小膜层的厚度, 导致检测出更多基底的物相信号.
层的a-PbO2的特征峰峰强更小, Hampson等[14]分析认为, 这是因为Cl−的存在加速了膜层的溶解, 导致膜层厚度减小. 值得注意的是, 在XRD谱中可以看到金属Pb的特征峰, 表明膜层不致密, 裸露出部分铅基合金基底. 还有一种解释是, 由于膜层厚度较薄, 导致X-射线穿透膜层, 检测到基底的信号. 含Cl−电解液中的膜层的Pb的特征峰更明显, 而且峰强更大. 这是由于Cl−的存在使膜层中有许多“火山口”状的孔洞, 降低了膜层的致密度, 使得更多的基底裸露. 此外, Cl−也可能减小膜层的厚度, 导致检测出更多基底的物相信号.
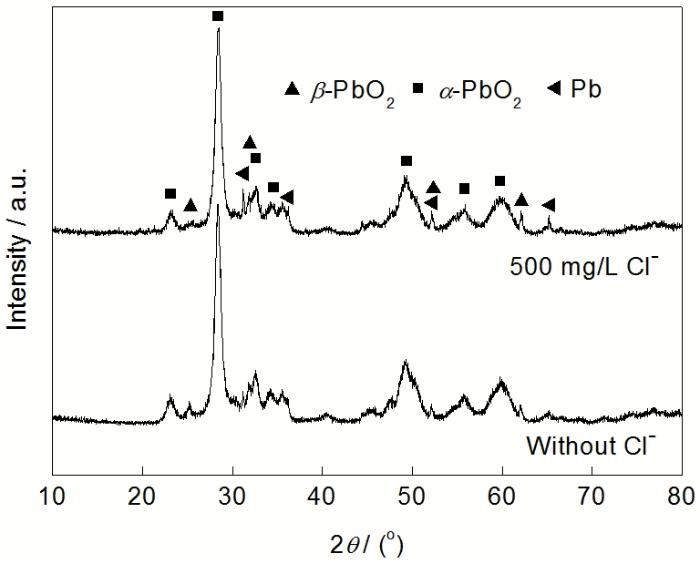
图3 Pb-Ag-RE阳极在无Cl-和含500 mg/L Cl- H2SO4溶液中恒流极化72 h后生成的氧化膜层的XRD谱
Fig.3 XRD patterns of anodic layers on Pb-Ag-RE anode formed through 72 h galvanostatic polarization in H2SO4 solution without Cl- and with 500 mg/L Cl-
2.3 腐蚀特性
2.3.1 Pb2+浓度 阳极在氧化腐蚀过程中, 首先以Pb2+的形式溶解在电解液中. 由于PbSO4的溶解度很小, 因此, Pb2+会与SO42−结合以PbSO4的形式在阳极表面沉积. 阳极表面的PbSO4会进一步氧化成PbO2, 最终导致膜层的形成. 此外, 溶液中溶解的Pb2+还会在电场的作用下, 向阴极迁移, 并还原沉积嵌入阴极产品中, 导致阴极产品的污染. 总的来说, Pb的腐蚀产物主要为阳极氧化膜层, 电解液中的Pb2+和阴极产品中的杂质Pb. 尽管电解液中Pb2+占Pb的腐蚀量比例很小, 但仍可以通过电解液中Pb2+浓度的变化来推测铅基合金的腐蚀情况. 图4所示的是恒流极化过程中2种电解液里Pb2+浓度随极化时间的变化. 在160 g/L H2SO4溶液中, 根据PbSO4的溶度积可算出Pb2+的饱和浓度约为0.078 mg/L(25 ℃, 标准大气压). 从图4可以看出, 含500 mg/L Cl−电解液中的Pb2+的浓度快速升高. 极化时间为36 h时, Pb2+的浓度达到0.09 mg/L (大于Pb的饱和浓度, 这与测试温  度高于25 ℃有关). 极化时间大于48 h后, Pb2+的浓度保持不变, 达到饱和. 而在无Cl−电解液中, Pb2+的浓度上升较慢, 极化持续72 h后, 其浓度才接近饱和. 因此, Cl−的存在会加快Pb2+在极化初期溶出, 加速合金基底腐蚀.
度高于25 ℃有关). 极化时间大于48 h后, Pb2+的浓度保持不变, 达到饱和. 而在无Cl−电解液中, Pb2+的浓度上升较慢, 极化持续72 h后, 其浓度才接近饱和. 因此, Cl−的存在会加快Pb2+在极化初期溶出, 加速合金基底腐蚀.
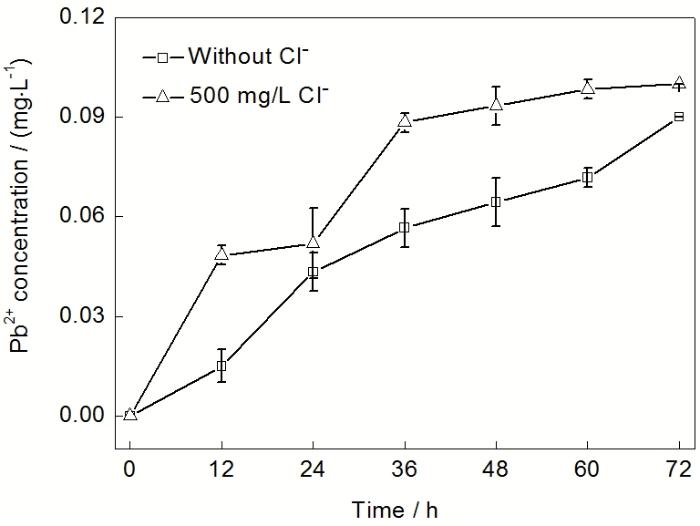
图4 Pb-Ag-RE阳极在无Cl-和含500 mg/L Cl- H2SO4溶液中恒流极化72 h过程中电解液中Pb2+浓度的变化
Fig.4 Variation of Pb2+ concentration in electrolyte during 72 h galvanostatic polarization in H2SO4 solution without Cl- and with 500 mg/L Cl-
2.3.2 腐蚀基底 铅基合金的腐蚀发生在合金基底/氧化膜层界面, 因此, 观察合金基底的腐蚀形貌有助于认识合金的腐蚀规律和特性. 在沸腾的糖碱溶液中浸泡5 min后, 氧化膜层溶解, 合金基底裸露出来. 尽管糖碱溶液在去除氧化膜层过程中也会对合金基底造成轻微的腐蚀, 但合金基底在恒流极化过程的腐蚀形貌基本可以保留下来, 如图5所示. 图5a是无Cl-电解液中Pb-Ag-RE阳极的基底腐蚀形貌. 从图5a可以看出, 基底较平整, 尽管有少量的腐蚀孔洞, 整体上腐蚀较均匀. 而在含500 mg/L Cl-的电解液中, 腐蚀基底不平整, 出现大量的腐蚀坑和孔洞, 表现出明显的局部腐蚀特征(图5b). 因此, Cl-的存在加剧了Pb-Ag-RE合金的点蚀. 图2b中在含Cl-电解液中, 氧化膜层有大量的像“火山口”状的孔洞, 这些孔洞的出现与基底的点蚀有关联. 在基底容易发生腐蚀的区域, 随着腐蚀产物的积累, 膜层内压增大并向外挤压, 从而导致膜层出现孔洞或裂纹. 在很多工程领域, Cl-都可以促进材料的腐蚀, 这主要是因为Cl-离子半径小、络合能力强. 在含Cl-电解液中形成的氧化膜层孔洞多, Cl-在膜层中的扩散和电迁移速度快, 进而导致Pb-Ag-RE合金基底的腐蚀加剧.

图5 Pb-Ag-RE阳极在无Cl−和含500 mg/LCl−H2SO4溶液中恒流极化72 h后基底的腐蚀形貌
Fig.5 Corrosion morphologies of the substrate of Pb-Ag-RE anode after 72 h galvanostatic polarization in H2SO4 solution without Cl− (a) and with 500 mg/LCl− (b)
2.4 析氧行为
铅基合金阳极在服役过程中主要进行析氧反应. 式(1)~(3)给出了被广泛接受的铅阳极在硫酸溶液中的析氧机理, 式中S代表氧化膜层表面的析氧活性位点[17]. 铅基合金阳极的析氧活性不仅仅影响阳极电位和电积能耗, 还对阳极的腐蚀有一定的影响. 因此, 有必要研究Cl-对Pb-Ag-RE合金阳极的析氧行为的影响. 本研究采用EIS和Tafel测试来研究析氧行为.
2.4.1 EIS Pb-Ag-RE阳极在2种电解液中极化72 h后测得的EIS如图6所示. 2种电解液中获得的复平面阻抗图均只有1个容抗弧, 对应 于双电层电容和传荷阻抗并联构成的RC回路. 在高频区, 出现感抗弧, 这是阳极表面分布不均匀的电活性物质发生电荷弛豫导致的[18]. 采用图6中给出的等效电路拟合阻抗数据, 拟合结果见图6中的曲线. 由于氧化膜层表面析氧活性位点分布不均匀, 拟合过程中采用常相位元件CPE来代替理想电容, 其阻抗ZCPE可以表示为[18]:
式中, Q为电容;
式中, Cdl是双电层电容, Ru是未补偿阻抗, Rct为传荷阻抗. 等效电路拟合获得Q, Ru, Rct及n值, 通过式(5)得到Cdl的数值, 具体结果见表1.
图6 所示EIS数据拟合后获得的阻抗参数
Impedance parameters determined by fitting the EIS data shown in Fig.6
| Electrolyte | L | Ru | Q | Cdl | n | Rct | χ2 |
|---|---|---|---|---|---|---|---|
| 10-7 H?cm2 | Ω?cm2 | 10-2 F?cm-2 | 10-2 F?cm-2 | Ω?cm2 | |||
| Without Cl− | 1.01 | 0.593 | 6.90 | 5.42 | 0.919 | 1.88 | 3.58×10−4 |
| 500 mg/L Cl− | 1.11 | 0.633 | 6.64 | 5.06 | 0.911 | 2.26 | 6.61×10−4 |
由表1可知, 2个EIS拟合结果的χ2值都在10−4数量级, 说明拟合的精确度符合要求. 从表1还可以看出, 在含Cl−电解液中, 未补偿阻抗Ru稍大于无Cl−电解液中的Ru. 这可能是由于在含Cl−电解液中生长的氧化膜层中有少量高阻抗(4×107~5×107 Ω?cm)的PbCl2嵌入. 含Cl−电解液中阳极的双电层电容Cdl值小于无Cl-电解液的Cdl, 说明在含Cl-电解液极化过程中, 氧化膜层/电解液界面上吸附有更少的析氧反应中间产物. 这是因为在含Cl-电解液中所生成的阳极氧化膜层表面PbO2的含量较低, 导致析氧活性位点更少. 此外, 从氧化膜层形貌可以看出, 含Cl-电解液中获得的膜层有些胶状区域, 导致表面积较小, 这也会导致其Cdl更小. 同时, 在含Cl-电解液中析氧反应传荷阻抗Rct的值更大. 因此, 结合更小的Cdl值和更大的Rct的值可以证明在含Cl-电解液中析氧反应更不易进行. 然而, Pb-Ag-RE阳极在含Cl−电解液中表现出更低的阳极电位. 这是因为, 铅基合金阳极的阳极电位是一种混合电位, 其不仅仅受析氧反应影响, 还受到其它副反应的影响, 如氯气的析出和基底的腐蚀等. 含Cl-电解液中更严重的基底腐蚀是阳极电位更低的一个解释, 因为腐蚀反应在更低的电位下进行, 起到减小阳极极化的作用.
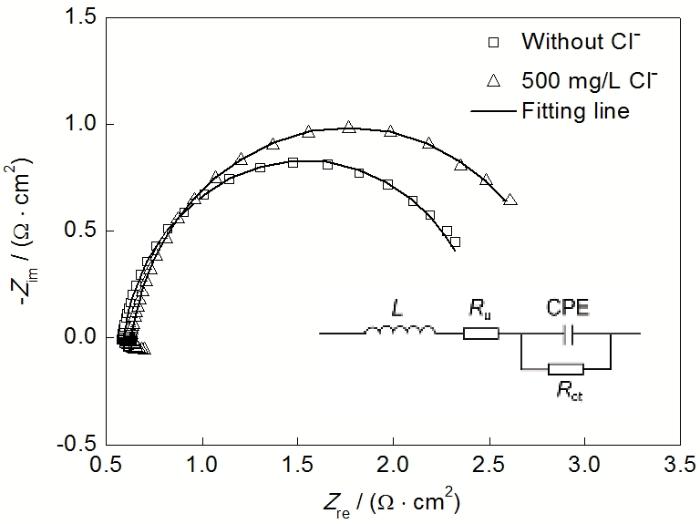
图6 Pb-Ag-RE阳极在无Cl−和含500 mg/L Cl− H溶 液中恒流极化72 h后的EIS和拟合过程采用的等 效电
Fig.6 EIS of Pb-Ag-RE anode after 72 h galvanostatic polarization in H2SO4 solution without Cl − and with 500 mg/L Cl− (Bias potential is 1.38 V in electrolyte without Cl− and 1.36 V in electrolyte with 500 mg/L Cl−) and electrical equivalent circuits (inset) used to fit the impedance data (L—inductance, Ru—uncompensated resistance, CPE—constant phase element, Rct—charge transfer resistance)
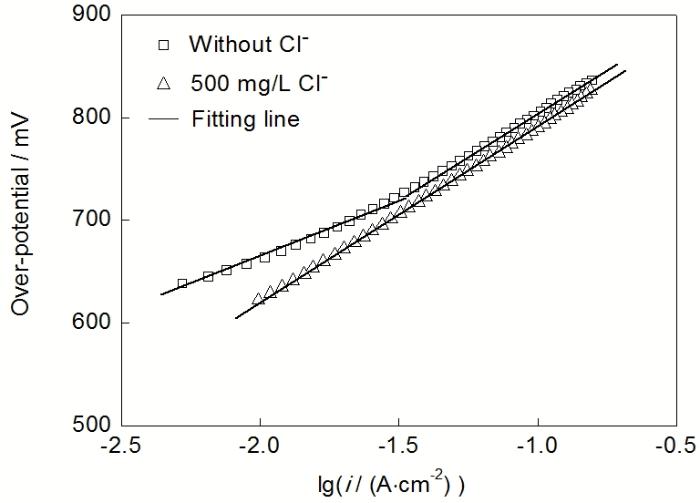
图7 Pb-Ag-RE阳极在无Cl−和含500 mg/L Cl− H2SO4溶 液中恒流极化72 h后的Tafel曲线
Fig.7 Tafel curves of Pb-Ag-RE anode after 72 h galvanostatic polarization in H2SO4 solution without Cl− and with 500 mg/LCl− (i—current density)
2.4.2 Tafel曲线 为了进一步研究Cl-对Pb-Ag-RE阳极析氧行为的影响, 在恒流极化72 h后进行了Tafel线性扫描测试. 图7显示的是由高电位向低电位准稳态电位扫描获得的Tafel曲线, 且所得Tafel曲线均按式(6)进行了修正[20]:
式中, Eappl为施加在阳极与参比电极之间的电压, i为电流密度, Eeff是扣除工作电极到参比电极之间溶液阻抗导致的压降iRu后阳极的实际电位. 由图7可见, 在无Cl-电解液中, Tafel曲线呈现双斜率特征. 采用Origin进行分段拟合后得到Tafel曲线的斜率. 在低过电位区间, Tafel曲线斜率为97.5 mV/dec. 在高过电位区间, Tafel斜率增加到163.7 mV/dec, 说明在此电位区间阳极反应受析氧中间产物的生成和吸附步骤控制. 在含500 mg/L Cl-电解液中, Tafel曲线只呈现单斜率特征(斜率为169.6 mV/dec), 说明Cl−对Pb-Ag-RE合金阳极的析氧反应机理有较大影响. 在低过电位区和高过电位区, 含Cl-电解液中析氧过程均由析氧中间产物的生成和吸附步骤控制. 在含Cl-电解液中, Cl-与水溶液的OH−和其他中间活性物质(阴离子或带负电的自由基)可能存在竞争吸附, 导致在氧化膜层/电解液界面吸附的析氧活性中间产物减少, 从而使其Tafel斜率较无Cl-电解液中的稍大, 这也与含Cl-电解液中Cdl值更小相对应. 综合EIS和Tafel的数据分析, 可以得出结论, Cl-的存在会抑制析氧中间产物的生成和吸附, 增加析氧传荷阻抗, 降低Pb-Ag-RE合金阳极的析氧活性.
3 结论
(1) 在含500 mg/L Cl- H2SO4溶液中, Pb-Ag-RE阳极在极化初期表现出更大的腐蚀速率. Pb-Ag-RE表面生长的氧化膜层出现“火山口”状的孔洞, 膜层致密度变差. 同时, 含Cl-电解液中合金基底点蚀严重. 因此Cl-会加速铅基合金基底的腐蚀.
(2) EIS和Tafel测试表明, 在含Cl-电解液中, 析氧反应中间产物的生成和吸附受抑制, 析氧传荷阻抗变大, 导致Pb-Ag-RE合金析氧活性更差. 但Cl-的存在使阳极腐蚀速率加快, 造成Pb-Ag-RE合金在含Cl−电解液中表现出较低的阳极电位.
(3) 500 mg/L Cl-对Pb-Ag-RE合金阳极腐蚀和析氧反应均有不利的影响. 工业生产中应尽量降低电解液中Cl−的浓度.
来源---金属学报 沪公网安备31011202020290号
沪公网安备31011202020290号
