



分享:镁基材料中储氢相及其界面与储氢性能的调控

李谦1,2,3, 孙璇3, 罗群3, 刘斌3, 吴成章3, 潘复生 ,1,2
,1,2
镁基储氢材料因储氢密度高、资源丰富、环境友好等优点而备受关注,但其存在吸/放氢温度过高、反应动力学缓慢和循环稳定性差等缺点,阻碍了其大规模产业化进程。尽管镁基储氢材料在新合金体系开发、纳米调控、催化修饰、多相复合等方面取得了巨大进展,但如何获取兼具吸/放氢容量高、温度适中、反应速率快及寿命长等优良性能的镁基储氢材料仍是一个挑战。本文较为系统地总结了镁基材料中储氢相及其界面的种类,论述了其微观组织/界面特征的调控策略和方法。重点探讨了储氢相组成、微观结构及其表/界面结构调控效果对提升储氢热力学与动力学性能的影响规律与作用机制,展望了通过调控储氢相及其界面来设计镁基储氢材料的前景和发展方向。
关键词:
氢能作为理想的二次清洁能源,兼具制取成本低、无毒无害、储量丰富等优点,以氢降碳有望成为实现“双碳”目标的重要举措之一。氢能的高密度安全储运是氢能全产业链中的重要环节。固态储氢材料因其安全、高效、高储氢密度等优势引起了广泛关注[1~4],其中,镁基材料因具有众多优点被认为是最有发展前景的储氢合金:① 镁资源丰富;② Mg密度低,仅为1.74 g/cm3;③ 高质量和高体积储氢密度(MgH2为7.6% (质量分数)和110 g/L);④ 优异的可逆性和环境友好性等[5~8]。但镁基储氢材料也存在活化性能差、热力学稳定性高和吸/放氢动力学缓慢的问题。如活化温度超过623 K,且需多次活化。Mg—H键需553~573 K的高脱附温度才能断裂,焓变值为75 kJ/mol H2[9]。H2分子/H原子在Mg/MgH2表面的解离/重组能力较差,H原子在Mg/MgH2中的扩散速率较低,导致吸/放氢速率缓慢[10~12]。针对上述问题,目前改善镁基储氢材料性能的主要策略有合金化、纳米/非晶化、催化剂掺杂以及先进的制备与加工等方法[13~16]。
储氢相是指在一定温度和H2压力下,能够储/放氢的金属或金属间化合物。Mg是镁基材料中可逆吸/放氢的最重要储氢相,通过合金化调控其相组成能加快吸/放氢速率,降低反应焓变[9]。在众多合金化元素中,过渡金属(TM)和稀土(RE)是与Mg合金化形成储氢相的常见元素,能够削弱Mg—H键,降低镁基氢化物的放氢温度[17,18],但合金化时引入非吸氢元素会降低储氢量。通过微合金化可调控镁基储氢相的种类、形貌和分布,能有效利用合金化生成第二相改善镁基储氢材料的综合性能。此外,制备方法也是调控储氢相及界面的常用技术手段,包括熔炼、粉末烧结、高能球磨(HEBM)、气相沉积、大塑性变形(SPD)等[13,19]。
镁基材料中储氢相的表/界面对H原子的吸附和扩散有着重要影响。与块体材料相比,粉末材料具有更高的比表面积,更利于氢的吸附[20]。镁基储氢材料中不同维度纳米结构的储氢相或氢化物相,因其独特的表面结构和成分分布,显著影响吸/放氢反应的热力学性质和动力学性能[21~23]。此外,材料内部储氢相的周期性结构在界面处中断,造成界面处化学键的不连续性,并在界面处形成畸变能,使界面具有不同于体相的特性,继而显著影响材料的储氢性能[24]。由于表/界面上H原子的吸附和扩散行为与体相结构大不相同,对镁基材料表/界面结构的调控有望大幅提升其吸/放氢性能。
本文基于不同镁基储氢材料的制备方法,归纳其对应的组织结构与界面特征,重点论述镁基材料中储氢相及其表/界面结构的调控效果对储氢性能的影响规律。在分析镁基储氢材料的国内外研究现状和发展动态的基础上,期望为设计具有优异储氢性能的镁基材料提供有价值的思考和借鉴。
1 镁基材料中的储氢相及其界面
1.1 主要储氢相及结构
Mg作为镁基材料中主要的可逆储氢相,为hcp晶体结构,可在约573 K和2.4~40 MPa氢压下与H2反应生成MgH2。当氢压较低时,形成低压相α-MgH2,反之则形成高压相β-MgH2和亚稳态相γ-MgH2[19]。
为改善MgH2的热力学性质,可通过非过渡金属、过渡金属和稀土金属等元素与Mg合金化形成可储放氢的二元/三元金属间化合物,如Mg-TM、Mg-RE、Mg-TM-RE体系等[19,25]。表1[19,25~41]总结了不同镁基储氢相的晶体结构和储氢容量。二元/三元镁基储氢相在初始吸/放氢之后会发生相分解,变成可逆储氢相(如Mg、Mg2Ni等)、非储氢相及催化相(如稀土氢化物REH x 等)。在Mg-TM二元体系中存在2种情况,一种是存在稳定的Mg-TM金属间化合物,最具代表性的是Mg2Ni,与H2在573 K和2 MPa下反应生成Mg2NiH4,其可逆储氢容量为3.6% (质量分数,下同)。其他Mg-TM金属间化合物(如Mg2Cu、Mg6Pd和Mg17Al12等)在氢化后没有形成三元氢化物,而是发生歧化反应形成MgH2及其他非储氢的金属间化合物[25]。另一种情况是不存在稳定的金属间化合物,可通过特定工艺形成Mg-TM-H系氢化物,如Mg-Fe-H和Mg-Co-H[19]。
表1 不同体系镁基储氢相的晶体结构和储氢容量[19,25~41]
Table 1
| Hydrogen | Pearson | Space group | Lattice | Hydride | Hydrogen storage | Ref. |
|---|---|---|---|---|---|---|
| storage | symbol | parameter | capacity | |||
| phase | nm | (mass fraction, %) | ||||
| Mg | hP2 | P63/mmc | a = b = 0.32, c = 0.52 | MgH2 | 7.60 | [19] |
| Mg2Ni | hP18 | P6222 | a = b = 0.52, c = 1.32 | Mg2NiH4 | 3.62 | [25] |
| Mg2Cu | oF48 | Fddd | a = 0.91, b = 1.8, | MgH2 | 2.53 | [25] |
| c = 0.53 | ||||||
| Mg17Al12 | cI58 |
|
bcc a = 1.06 | MgH2 | 4.40 | [19] |
| Mg3La | cF16 |
|
bcc a = 0.75 | MgH2, LaH x (x = 2-3) | 2.89 | [26] |
| Mg17La2 | hP38 | P63/mmc | a = b = 1.04, | MgH2, LaH x (x = 2-3) | 4.87 | [27,38] |
| c =1.02 | ||||||
| Mg12Ce | tI26 | I4/mmm | a = b = 1.03, c = 0.60 | MgH2, CeH2.73 | 5.68 | [28] |
| Mg3Ce | cF16 |
|
bcc a = 0.74 | MgH2, CeH2.73 | 3.62 | [28] |
| Mg12Nd | tI26 | I4/mmm | a = b = 1.03, c = 0.59 | MgH2, NdH2.61 | 5.63 | [29] |
| Mg24Y5 | cI58 |
|
bcc a = 1.13 | MgH2, YH x (x = 2-3) | 5.34 | [30] |
|
18R-LPSO (Mg85Zn6Y9) |
- | C2/m | a = 1.11, b = 1.93, | MgH2, YH x (x = 2-3) | 4.60-5.30 | [31] |
| c = 1.60 | ||||||
| 18R-LPSO | - | P6322 | a = b = 0.31, c = 4.86 | MgH2, Mg2NiH4, YH x | 4.60 | [32] |
| (Mg12YNi) | (x = 2-3) | |||||
| 14H-LPSO | - | P63/mcm | a = b = 1.11, c = 3.66 | MgH2, YH x (x = 2-3) | 5.48 | [33] |
| (Mg92Cu3.5Y4.5) | ||||||
| Nd4Mg80Ni8 | tI92 | I41/amd | a = b = 1.27, c = 1.59 | MgH2, Mg2NiH4, NdH2.61 | 4.94 | [37] |
| Nd16Mg96Ni12 | oS124 | Cmc21 | a = 1.53, b = 2.17, | MgH2, Mg2NiH4, NdH2.61 | 3.92 | [36] |
| c = 0.95 | ||||||
| NdMg2Ni | oS16 | Cmcm | a = 0.41, b = 1.02, | MgH2, Nd2.61 | 2.07 | [39] |
| c = 0.83 | ||||||
| LaMg2Ni | oS16 | Cmcm | a = 0.42, b = 1.03, | LaMg2NiH7 | 1.92 | [39] |
| c = 0.84 | ||||||
| La2MgNi9 | hR36 |
|
a = b = 0.50, c = 2.43 | La2MgNi9H12 | 1.60 | [40] |
| LaMgNi4 | cF24 |
|
bcc a =0.72 | LaMgNi4H7 | 1.40 | [41] |
| 2H-La3MgNi14 | - | P63/mmc | a = b = 0.50, c = 2.42 | La3MgNi14H18 | - | [34] |
| 3R-La3MgNi14 | - |
|
a = b = 0.50, c = 3.61 | |||
| 2H-La4MgNi19 | - | P63/mmc | a = b = 0.50, c = 3.23 | La4MgNi19H24 | 1.50 | [35] |
| 3R-La4MgNi19 | - |
|
a = b = 0.50, c = 4.82 |
Mg与稀土元素(Y、La、Ce、Pr、Nd、Sm和Gd)可形成REMg12、RE2Mg17、REMg3、REMg2和REMg等一系列相对稳定的二元储氢相[26~30]。RE元素含量的变化会导致Mg-RE系储氢相结构的变化。以RE2Mg17和REMg3为例,RE2Mg17具有Th2Ni17结构,空间群为P63/mmc,在Mg-La/Ce体系中较为稳定,而REMg3具有BiF3结构,在Mg-La/Ce/Pr/Nd/Sm/Gd体系中较为稳定[24]。Mg-RE系中二元储氢相氢化后会分解为MgH2和催化相REH x。由于稀土氢化物的分解平台较低,在一定的温度范围内很难分解,Mg-RE系储氢相的可逆储氢容量主要由MgH2贡献。
Mg-TM-RE (TM = Zn、Cu、Ni)体系中可以形成多种三元储氢相,如长周期堆垛有序结构(LPSO)相和超晶格点阵相[31~35,42]。LPSO相呈现高含量的Mg以及均匀分散的TM和RE元素,表现出良好的储氢能力。在Mg-TM (Zn, Cu, Ni)-RE (Y, Gd, Ce)合金中的LPSO相包括24R、14H、18R、10H和12R等[43]。其中,Mg-Ni-RE体系中的LPSO相在第一次吸/放氢后会原位分解形成储氢相Mg和Mg2Ni及催化相REH x,REH x 在吸/放循环过程中均匀分散在Mg基体中,表现出优异的催化活性[33,44~46]。此外,REH x 起到钉扎Mg/MgH2相界的作用,抑制其晶粒生长,增强镁基合金的结构稳定性[47]。例如,具有18R-LPSO相的Mg12NiY合金在573 K下的储氢容量达到4.6%[32],具有14H-LPSO相的Mg92Cu3.5Y4.5合金储氢容量达到5.48%[33]。Mg-Ni-Nd体系中没有LPSO相,但富Mg角存在多种新的三元储氢相,如Nd4Mg80Ni8、Nd16Mg96Ni12、NdMgNi4、NdMg2Ni等[36,48],其中Nd4Mg80Ni8和Nd16Mg96Ni12的储氢容量分别为4.94%和3.92%,且Nd4Mg80Ni8相具有优异的循环寿命,在573 K下819 cyc循环后仍保持最高储氢容量的80%[37]。
RE-Mg-Ni超晶格金属间化合物主要是在RENi3、RE2Ni7和RE5Ni19的基础上发展起来的三元储氢相,典型代表有La2MgNi9、La3MgNi14和La4MgNi19[49]。RE-Mg-Ni超晶格金属间化合物具有层状结构,即[REMgNi4]和[RENi5]单元沿着c轴方向以一定的方式组合形成一个复杂单元,并且2个或2个以上这种复杂单元同样沿着c轴方向有序堆垛形成一个周期晶胞,其化学式可以表达为m[REMgNi4]·n[RENi5],即RE n + m Mg m Ni5n + 4m,其中m和n分别为每个复杂单元中[REMgNi4]和[RENi5]的数量。根据不同的单元堆积方式,RE-Mg-Ni超晶格金属间化合物有2种基本晶体结构类型,即2H相和3R相。由于RE-Mg-Ni三元储氢相中的Mg含量较低,导致其储氢量不足2%[34,35]。
1.2 镁基储氢相的界面
镁基材料的表/界面结构和组成分布可直接影响H原子的吸附和扩散,进而作用于材料的储氢性能。在镁基储氢材料的设计中,了解表/界面结构对吸/放氢反应的影响,有助于通过合理调控界面结构来提升其储氢性能。储氢材料吸/放氢反应过程涉及到的界面主要为气-固界面和固-固界面。气-固界面主要是指H2与材料接触的外表面结构,在表界面设计和构筑过程中需要考虑比表面积、表面取向和表面能等几个重要因素。例如球磨法制备的镁基材料粉末相比块状材料具有较大的比表面积,从而使其具有更高的反应活性。H2在表面的吸附能与表面取向有关,如Pd(110)和Pd(111)表面的吸附能分别为24.4和20.8 kJ/mol[50]。材料表面的缺陷同样会影响H2吸附性能。例如,Mg(0001)表面的空位缺陷有利于氢解离过程,能垒为1.28 eV,小于无缺陷表面的1.42 eV[51]。在评价储氢性能指标中,活化、抗氧化/粉化能力以及循环寿命等均与表面结构密切相关。有效调控表面结构的手段主要有减小颗粒尺寸、增大粗糙度、制备一维纳米线/二维纳米薄膜结构、合成中空、多孔或介孔的框架结构等。
固-固界面主要可以概括为2种:一种是结构相同而取向不同晶体之间的界面,如晶界、亚晶界,其他如孪晶界等则属于特殊晶界。通过SPD、熔体快淬(MS)或放电等离子烧结(SPS)制备的块体细晶材料,在材料内部形成高密度的大角度晶界,能够增加H原子传输通道[52]。另一种是结构、取向以及化学成分都不相同的晶体之间的界面,可利用合金化手段在镁基材料中引入多种共晶相,从而丰富相界面,这其中包括储氢相之间和储氢相与非储氢相之间的界面,吸/放氢过程中形成的Mg-MgH2相界及镁基储氢相-催化相界面等[53,54]。氢扩散与氢化物形核主要取决于氢化物-储氢相、储氢相-储氢相、储氢相-非储氢相及储氢相-催化相之间的界面。类似于晶界或相界,层错界面也是与储氢动力学性能息息相关的有效界面[55]。
2 镁基储氢材料的制备与储氢相及其界面的调控
2.1 物理制备方法
2.1.1 物理工艺辅助熔炼
高温熔炼作为制备镁基储氢合金传统且常见的工艺,可实现大规模生产,但常规熔炼制备合金中α-Mg相和其他相尺寸过大且分布不均。超声处理(UST)作为一种环境友好且成本低的物理工艺,已应用于各种镁基合金的熔体加工。Ding等[56]通过UST改变Mg-Ni亚共晶凝固过程中组成相的形貌、尺寸和分布。经UST处理后,α-Mg相由铸态的花瓣状逐渐球化,直径为160~180 µm,且Mg-Mg2Ni共晶相明显细化。超声的空化与声流破碎镁基合金凝固过程中形成的树枝晶,阻碍镁合金晶粒长大,使得镁基合金中α-Mg相和其他物相显著球化、细化和均匀化,提高了合金晶界/相界分数。
超高压(SHP)技术也被用于辅助熔炼,旨在细化材料中的物相尺寸,改变相的形貌与组成,提高其界面结构分数。Sun等[45]利用SHP技术处理Mg12NiY合金,可使Mg2Ni相向亚稳相Mg6Ni转变,且各相尺寸更加细小,界面分数增加,尤其是18R-LPSO相宽度尺寸由铸态的5~15 μm减小至0.05~0.2 μm,体积占比也有所提升。Fu等[57]通过SHP技术制备Mg-5%Ni合金,同样存在共晶相Mg2Ni向亚稳相Mg6Ni的物相转变;不同的是,Mg-5%Ni合金获得了小角度取向及高指数晶面的枝晶界面结构,枝晶界面分数随SHP处理温度的提高而增加。
2.1.2 熔体快淬
典型的熔体快淬是将熔化的金属或合金在Ar气保护下,喷射到高速旋转的辊轮表面,熔融材料与冷却辊轮表面接触后,在1 × 105 K/s的高冷却速率下迅速凝固,形成非晶态或非晶/纳米晶复合结构的过程。熔体快淬制备的Mg-Ni-RE三元合金已被广泛用于储氢研究[58],但要获得完全非晶态Mg-Ni-RE合金是不容易的。在Mg-Ni-Ce三元体系中,Ce比Ni更有利于富镁Mg-Ni-Ce体系的非晶态形成。Ce含量达到4%,Mg-Ni-Ce合金发生部分非晶化,当Ce含量达到5%,可以获得完全非晶结构[59]。
2.1.3 粉末冶金
粉末冶金具有反应温度低的优势,可克服高温熔炼法中Mg易挥发和合金成分不易控制等缺点。Knotek等[60]通过放电等离子烧结方法获得孔隙率为20.6%~24.3%的Mg-Ni基合金,其微观组织由球状亚微米颗粒组成。Song等[61]采用SPS方法合成Mg-V77.8Zr7.4Ti7.4Ni7.4和Mg-V38Zr25Ti15Ni22镁基复合材料,在Mg相和钒基固溶体之间形成薄的纳米Mg相过渡区。与常规烧结方法相比,SPS技术提供更多控制烧结的可变参数,可以获得具有亚稳态微观结构和高孔隙率的镁基复合材料。
激光烧结可在快速加热和冷却过程中引入大量的晶体缺陷、微裂纹以及细晶。Liu等[62]通过激光烧结制备Mg-20%LaNi5复合材料,物相组成为Mg、Mg2Ni和LaMg12。网状共晶Mg + Mg2Ni和块状LaMg12组成的网格包裹Mg晶粒,呈核/壳组织结构,可有效改善镁基材料的氢化与电化学性能。
2.1.4 球磨
球磨法能够制备镁纳米晶材料、Mg与催化剂或其他储氢材料的复合材料,可有效地减小颗粒与晶粒尺寸、增加比表面积,并引入大量的晶界和晶格畸变,改善材料在气相、化学溶液或电解质中的化学反应活性[19,58,63,64]。为了提高球磨效率,超声波及介质阻挡放电等离子体等物理装置被用于辅助高能球磨。例如,超声波辅助高能球磨同时对粉末进行超声波、机械力和声空化作用, 从而加速破碎,促进扩散,实现固相反应。介质阻挡放电等离子体辅助球磨工艺将等离子体场和机械球磨结合,促使粉末颗粒细化及表面改性,获得独特的组织结构。同时,能够加速原位固相反应、气固反应和机械合金化进程。值得一提的是,该方法解决了Mg(In)和Mg(In, Al)固溶体的制备问题,提高了制备效率[58]。
2.1.5 大塑性变形
大塑性变形可获得超细晶粒尺寸和具有大比例晶格缺陷的块体镁基材料,是一种改善材料储氢性能的方法,其中包括高压扭转(HPT)、等通道转角挤压(ECAP)、冷轧(CR)和锻造等工艺。
HPT方法能够制备圆盘状超细晶(UFG)储氢材料,其表面的元素偏析与裂纹可促进吸氢活化,提高空气抗失活率;引入的晶界、堆垛层错和位错等晶格缺陷,可充当H原子快速扩散的路径,增强吸/放氢动力学和活化性能[55,65,66]。HPT对控制固态相变、合成新合金及低温下合成金属间化合物是非常有效的,被应用于合成新型的镁基储氢材料[67~69]。Edalati等[70]采用第一性原理计算设计了一种具有bcc基CsCl型结构的Mg4NiPd合金,通过HPT工艺加工1500道次后制备出该合金,其元素分布、晶体结构及微观组织如图1[70]所示。合金的3种元素在原子水平上均匀分布,透射电镜(TEM)像表明Mg4NiPd合金为独特的纳米结构,平均晶粒尺寸为(10 ± 4) nm,具有扭曲的多晶结构。
图1
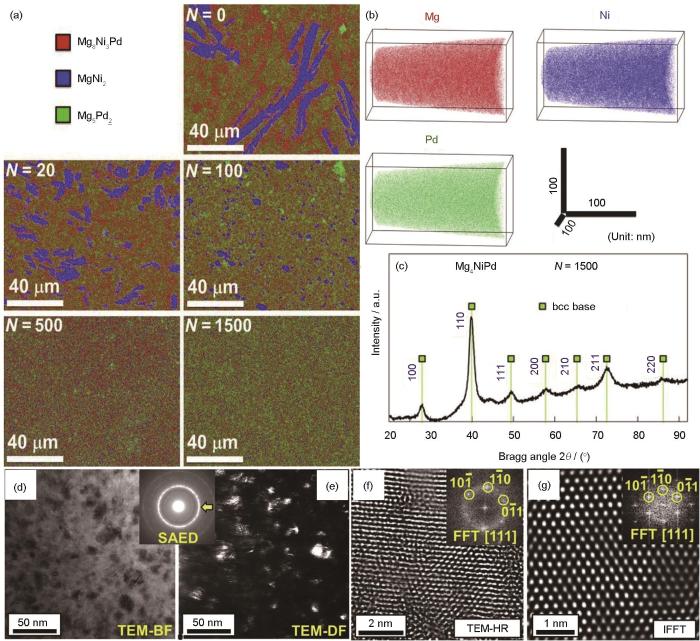
图1 高压扭转(HPT)处理0~1500道次Mg4NiPd的SEM-EDS图,及HPT处理1500道次Mg4NiPd的元素分布、XRD谱和微观组织TEM分析[70]
Fig.1 SEM-EDS and TEM analyses of Mg4NiPd treated by high pressure torsion (HPT)[70]
(a) elemental map of Mg4NiPd treated by HPT for 0-1500 turns (N) (b, c) atom probe tomography (APT) elemental map (b) and XRD spectrum (c) of 1500 turns of Mg4NiPd (d) bright-field TEM image (e) dark-field image and corresponding selected area electron diffraction (SAED) pattern (f) high-resolution TEM image and corresponding fast Fourier transform (FFT) pattern (inset) (g) lattice image of {110} diffraction reconstructed by inverse FFT (IFFT) (inset)
ECAP能有效地细化晶粒,产生空位、位错等晶体缺陷,并在镁基材料中引入大量的高角度晶界和择优取向。由于材料结构受通道角度、路径、道次、挤压速率、挤压温度等多个工艺参数的影响,为调控镁基材料微观结构提供较大可操作空间[71,72]。冷轧也是储氢领域中常被采用的加工方法,尽管其细化效果不如ECAP工艺,但采用冷轧方法加工具有(0001)择优取向的镁基材料更具有工业应用潜力。通过将ECAP与CR工艺结合能够实现微纳米晶粒尺寸与(0001)择优取向共存的组织特征[73]。锻造工艺具有成本低及工业化加工金属材料的优势,在储氢领域也有所应用。锻造MgH2的吸氢动力学与CR或HPT制备样品相似,但随着锻造道次的增加,大量的MgO随之形成。为避免这一问题,研制了Ar气氛下单道次高变形速率锻造技术,可用来制备具有择优取向与大量缺陷共存的储氢材料[74]。
2.1.6 物理气相沉积法
氢等离子体金属反应 (HPMR)已成功用于制备纯Mg和镁基金属间化合物的纳米颗粒,可通过控制氢压和电流调节粒径。Shao等[22]通过HPMR技术制备出高纯度纳米Mg颗粒,由于Mg的蒸气压较高,Mg颗粒的平均直径超过300 nm。研究者相继制备了Mg-Ni、Mg-Co和Mg-Cu等镁基纳米复合材料以期得到更小的颗粒尺寸[22]。图2a~c[22]为HPMR制备的Mg2Ni、Mg2Co和Mg2Cu纳米颗粒。
图2

图2 氢等离子体金属反应(HPMR)法制备的镁基纳米颗粒,磁控溅射制备Mg-Nb合金薄膜,及气相反应合成的Mg纳米线SEM像[21,22,75]
Fig.2 Magnesium-based nanoparticles of Mg2Ni (a), Mg2Co (b), and Mg2Cu (c) prepared by hydrogen plasma-metal reaction (HPMR) method[22]; surface and cross-sectional (insets) morphologies of Mg0.83Nb0.17 (d), Mg0.70Nb0.30 (e), and Mg0.24Nb0.76 (f) prepared by Co-sputtering[75]; and SEM images of Mg nanowire synthesized by gas phase reaction at flow rates of 200 cm3/min (g), 300 cm3/min (h), and 400 cm3/min (i)[21]
磁控溅射和热蒸发技术为镁基储氢材料的纳米结构形态、成分和界面特性的精确控制提供了一种有效途径。磁控溅射法制备的Mg/Pd多层膜由细晶Pd层和柱状晶Mg层组成,通过控制溅射功率来调节溅射原子的速率,形成具有不同界面结构及组织的Pd/Mg薄膜。图2d~f[75]为磁控溅射方法制备的具有bcc结构的Mg-Nb合金薄膜的表面与截面形貌,不同Nb含量的薄膜显现不同的表面形貌。通过气相沉积实现了一维纳米线Mg/MgH2结构的可控制造,其中氢被用作反应性气体以直接产生纳米结构MgH2,较高的H2压力下为更规则的针状纤维,而较低的H2压力下为随机弯曲的纳米线[21],不同流量下气相反应合成Mg纳米线如图2g~i[21]所示。
2.2 化学制备方法
2.2.1 化学还原法
化学还原法是利用氧化还原反应在液相中将金属离子还原成金属原子,制备出纳米金属粒子的方法[76],其中Rieke法最为成功。Liu等[77]通过Rieke方法,在高锂/萘比例下的四氢呋喃(THF)溶液中共沉淀产生了8 nm的Mg颗粒,后又通过改良的Rieke法合成Mg-X (X = Fe、Co、V)纳米复合材料,由不规则形状的颗粒聚集在一起组成,粒径范围为10~20 nm[77]。化学还原法还可用来制备具有核/壳结构的镁基纳米复合材料,有研究者将Mg粉与THF溶液中的TMCl x 反应,制备了一系列Mg-TM (TM = Ti、Nb、V、Co、Mo和Ni)核/壳纳米复合材料,TM壳层厚度小于10 nm[78]。Cho等[79]基于简单溶液的化学还原方法合成了一种新型的、环境稳定的rGO-Mg纳米复合储氢材料,还原氧化石墨烯片(rGO)包裹Mg纳米晶体可免受O2和湿气的影响。化学还原法不需要外界电源、操作简单易行,可在Mg基体表面均匀包覆催化相且包覆层厚度可控。
2.2.2 化学气相沉积
通过氢化化学气相沉积(HCVD)的气固反应[17],可以合成具有局部涂敷超薄MgO层的单分散Mg2NiH4单晶纳米颗粒,其HCVD合成过程分为4个步骤:将Mg蒸气输送到Ni/石墨烯片(graphene sheets,GS)衬底上; Ni掺杂剂在GS衬底上原位形成单分散的Mg2Ni纳米颗粒; Mg2Ni纳米颗粒吸氢转化为Mg2NiH4纳米颗粒;在自然冷却过程中引入Ar/空气混合气体,经钝化后,在Mg2NiH4纳米颗粒表面局部涂敷MgO层。
2.2.3 机械合金化
目前,采用机械合金化已成功合成Mg-Ni、Mg-Co等二元合金及Mg-Ni-RE (RE = Nd、Ce、Pr等)三元合金。机械合金化在成分和球磨时间适当时,会形成元素均匀分布的纳米/非晶结构[80]。如球磨60 h的REMg11Ni (RE = Nd[81]、Ce[82]、Pr[83]) + x%Ni (x = 100或200)合金呈现出纳米晶与非晶混合结构,并具有位错、晶界和孪晶等结构缺陷。另外,通过长时间的反应球磨Mg-Co[84]二元体系及Mg-V-TM (TM = Ni、Co、Cu)[85]、Mg-Ca-V[86]三元体系能够合成具有bcc晶体结构的镁基储氢材料。此外,在一定H2压力下通过反应球磨合成镁基氢化物的方法,有效地避免了Mg及镁基合金长时间研磨过程中的氧化问题,提高了氢化物纯度及产率。在Mg-Fe和Mg-Co等体系中不存在稳定的二元金属间化合物,通过氢化反应球磨可成功制备Mg2FeH6和Mg2CoH5三元氢化物[19]。
表2[21,22,56~62,70~73,77~79,81~83]总结了镁基储氢材料的不同制备方法对储氢相、组织结构和表/界面等特征的影响,制备工艺参数的变化可极大地改变储氢相及其界面,直接作用于镁基材料储氢热力学与动力学性能。
表2 不同调控方法下镁基材料储氢相及组织/界面对比[21,22,56~62,70~73,77~79,81~83]
Table 2
| Regulatory | Sample | Hydrogen storage | Particle | Microstructural characteristic | Ref. |
|---|---|---|---|---|---|
| method | status | phase | grain size | ||
| Smelting | Bulk | Mg, Mg-TM, | Several | Large grain/phase size, limited interface fraction | [56,57] |
| Mg-RE, | hundreds of | ||||
| Mg-TM-RE | microns | ||||
| Melt-spinning | Ribbon | Mg-Ni, | - | Amorphous or nano/amorphous composite structure, interfaces between amorphous and nanocrystalline | [58,59] |
| Mg-Ni-RE | |||||
| Powder | Bulk | Mg-based | Tens of | High porosity, core/shell structure, plenty of vacancies and cracks defect, phase/grain boundary | [60-62] |
| metallurgy | composite phase | microns | |||
| Severe | Bulk | Mg-based | Micro/nano | Ultra-fine grain, high density grain/phase boundaries, twin boundaries, dislocation, stacking fault, texture | [70-73] |
| plastic | metastable | grain size | |||
| deformation | phase | ||||
| Vapor | Powder/ | Mg, Mg-TM, | 20-600 nm | Nano-particles, nano-films, nano-wires, surface structure, surface orientation, interfacial energy, interfacial stress | [21,22] |
| deposition | thin film/ | Mg-Al, Mg-V, | |||
| wire | Mg-Y | ||||
| Chemical | Powder | Mg, Mg-TM | 10-20 nm | Nano-particles, core/shell structure, high purity, high specific surface area | [77-79] |
| reduction | |||||
| Mechanical | Powder | Mg, Mg-TM, | Micro/nano size | Nano/amorphous composite structure, bcc crystal structure, crystal defect, metastable structural interface | [81-83] |
| alloying | Mg-RE, | ||||
| Mg-TM-RE |
3 镁基材料中储氢相及其界面特征与储氢性能的相关性
3.1 储氢相对储氢性能的影响
3.1.1 相组成的影响
通过熔炼、粉末冶金和机械合金化等工艺合成二元或三元镁基储氢相可以实现调控镁基储氢材料的吸/放氢热力学和动力学性能。与纯Mg相比,已有报道的Mg-TM体系,包括Mg-Cu、Mg-Ni、Mg-Co、Mg-Ti、Mg-Pd和Mg-Fe等二元体系表现出更好的吸/放氢热力学和动力学性能,这是由于Mg—H键通过H的电子与过渡金属中不饱和d/f壳层电子之间的相互作用效应而减弱[9]。部分Mg-TM体系氢化后形成了萤石晶体结构MgTMH y,其H2迁移率明显高于金红石结构的MgH2,改善了吸/放氢速率[87]。
Mg和RE元素形成的Mg-RE二元储氢相也表现出储氢动力学性能的明显提升,原位形成的稀土氢化物(REH x )引入更多的界面通道,促进氢在合金中的扩散。有研究[88]报道REH x 具有独特的“氢泵”功能,可有效降低表观活化能。Mg-Ni-RE三元储氢相氢化分解形成的多相复合材料可提高动力学及循环性能。如Mg80Ce18Ni2合金可逆储氢容量可达4.0%以上,放氢速率比氢化的Mg3Ce合金要快,Mg80Ce18Ni2合金吸/放氢循环温度显著降低至505 K,比纯Mg降低约100 K,并且在500 cyc循环后,氢容量可保持在80%以上[88]。Mg80Ce18Ni2合金储氢性能的提升在于氢化分解后原位形成的CeH2.73-MgH2-Ni复合材料,Ni与CeH2.73起到了良好的催化作用。在Mg80Ce18Ni2合金首次氢化过程中,Mg3Ce相先与H2反应生成CeH2.73-Mg,然后CeMgNi4相氢化分解在CeH2.73-Mg的边界处形成Ni,使得合金转变为Mg-CeH2.73-Ni复合材料后,Mg与H反应生成MgH2。
在Mg-Ni-RE (RE = Y、Nd)三元体系中[37,89],Nd4.3Mg87.0Ni8.7合金经氢诱导分解为NdH2.61-Mg-Mg2Ni复合材料,其最大可逆储氢量为4.77%,经819 cyc循环后仍可吸氢3.75%,容量保留率为78.6%[37]。具有18R-LPSO结构的Mg86.1Ni7.2Y6.8三元储氢相经氢诱导分解可形成Mg-Mg2Ni-YH2复合材料,其最大可逆储氢量为5.2%,620 cyc循环后仍保有4.3%储氢量[89]。Mg-Ni-RE体系中氢致诱导形成的复合材料具有良好动力学性能与循环稳定性的原因有以下3点:① 原位形成的REH x -Mg-Mg2Ni提供大量相界与晶界;② REH x 和Mg2Ni对Mg的催化作用;③ REH x 分散在Mg和Mg2Ni基体中起到钉扎作用,保证储氢相微观结构的良好稳定性。
HPT加工过程中严重的剪切应变、高压以及晶格缺陷诱导的原子扩散,使该方法成为控制相变的有效工具。HPT工艺与热处理相结合,对于从不混溶的单质粉末混合物中合成新的镁基亚稳相是非常有效的。表3[65,67~70]总结了HPT制备得到的镁基亚稳相。在这些镁基亚稳相中,部分氢化后会分解形成MgH2及其他金属氢化物,但一些亚稳相吸氢后并未形成氢化物相,吸氢后的晶体结构与HPT处理后基本相同。有研究[67]表明,未形成氢化物相且能快速吸/放氢的原因为,H原子被镁基纳米团簇所吸收,HPT处理引入的大部分晶界作为样品表面和纳米团簇之间氢传输的途径,提高了吸放/氢速率。Edalati等[70]设计了具有接近零的低氢结合能的新型三元镁基化合物,该合金在室温下可逆吸/放氢量为0.7%,其加工策略是通过HPT工艺在原子尺度上混合2种不同类型的二元化合物:与H结合能为负的Mg2Ni和与H结合能为正的Mg2Pd。
Table 3
| Metastable phase | Lattice parameter / nm | Decomposition | Hydride | Hydrogen storage | Ref. |
|---|---|---|---|---|---|
| temperature | performance | ||||
| K | (mass fraction) | ||||
| Mg2Ti |
bcc a = 0.345 fcc a = 0.429 hcp-I a = 0.319, c = 0.517 hcp-II a = 0.297, c = 0.514 |
643 | MgH2, | - | [68] |
| TH2 | |||||
|
Mg-50%Zr (atomic fraction) |
bcc a = 0.3566 fcc a = 0.44-0.46 hcp a = 0.321, c = 0.516 |
> 773 |
- |
Absorb: 1.0% (20 s, 303 K, 9 MPa) |
[67] |
| MgVCr | bcc a = 0.295 | > 573 | - | Absorb 0.9% H2 and the reversibility is 0.4% at 303 K | [69] |
| MgV2Cr | bcc a = 0.294 | 573 | MgH2 | ||
| Mg-V-Sn | b2-type structure a = 0.360 | - | - | - | [65] |
| Mg-V-Pd | b2-type structure a = 0.316 | - | - | - | |
| Mg-V-Ni | bcc a = 0.325 | - | - | - | |
| Mg-Ni-Pd | bcc-based CsCl-type structure | 440 | - | 0.7% reversible hydrogen | [70] |
| a = 0.319 | absorption and desorption | ||||
| at 305 K |
3.1.2 晶体结构的影响
具有bcc结构的储氢合金被认为是更有吸引力的储氢材料,其原因在于:① 具有bcc结构的金属,H占据四面体和八面体间隙位点的数量是fcc或hcp结构的3倍;② H在bcc晶格中的扩散通常比fcc和hcp晶格中的快几个数量级[84]。早期研究[85]通过机械合金化成功合成新型bcc结构Mg-TM-V (TM = Ni、Co、Cu)三元合金,它们在3 MPa和298 K下的吸氢量分别为2.4%、1.44%和0.95%。
在0.1 MPa的Ar气下以200 r/min的转速球磨Mg、Co粉混合物0.5~400 h,合成的bcc结构Mg-Co合金能够在低于室温的情况下吸氢而无需任何活化过程[84]。图3[84]为球磨制备Mg55Co45合金的微观组织与压力-成分-温度(PCT)曲线。球磨125 h获得Mg55Co45合金颗粒尺寸为1~3 μm,TEM暗场像表明合成的Mg55Co45合金晶粒尺寸在几纳米左右。据Rietveld分析,bcc结构晶格常数为0.30092 nm。该合金即使在258 K下也可吸氢3.24%,其氢化物仍然是bcc结构。值得注意的是,具有0.300~0.305 nm范围内晶格参数的bcc结构,如Mg-V-TM (TM = Ni、Co、Cu)[85]和Mg-Ca-V[86],都可以在室温下吸氢。机械合金化引入的结构缺陷和具有合适晶格参数的bcc结构是室温下吸氢动力学性能增强的主要原因。
图3
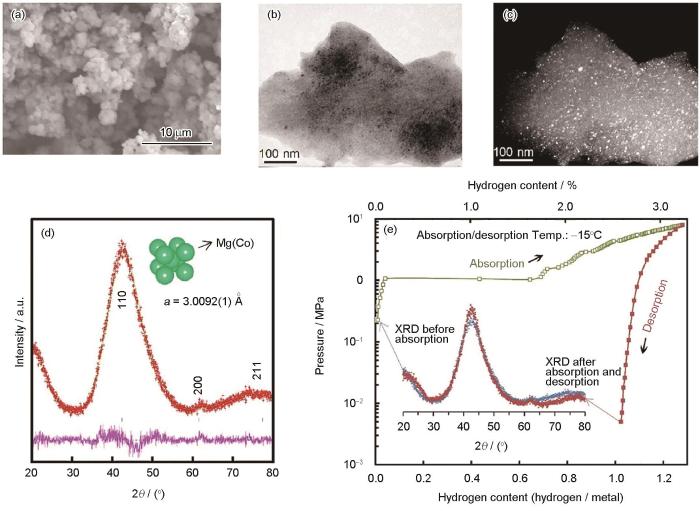
图3 Mg55Co45合金样品的SEM和TEM像、XRD谱及258 K时Mg55Co45 bcc合金的压力-成分-温度(PCT)曲线[84]
Fig.3 SEM image (a), TEM images (b, c), and XRD spectra (d) of Mg55Co45 alloy sample, and pressure-composition-isotherm (PCT) curves of the Mg55Co45 bcc alloy at 258 K (Inset: XRD curves of the Mg55Co45 bcc alloy before/after absorption and desorption cycle) (e)[84]
3.1.3 纳米结构的优势
构建纳米结构的镁基材料是提高Mg/MgH2体系储氢热力学与动力学性能的有效方法。Shao等[22]通过HPMR制备了平均颗粒尺寸为300 nm的纯Mg颗粒,材料表现出高储氢量与良好的动力学,在高于573 K的温度下30 min内可吸氢7.5%,MgH2形成焓为-75.0 kJ/mol H2。有计算研究表明,MgH2的热力学稳定性随颗粒粒径的减小而降低,但在尺寸上存在一个阈值,通过第一性原理计算得到MgH2颗粒尺寸低于1.3 nm时,分解焓会显著降低。如MgH2颗粒尺寸为0.9 nm时,其解吸焓降低至63.0 kJ/mol H2,解吸温度仅为473 K[90]。Zhang等[91]在THF溶液中利用超声驱动的液-固置换反应成功合成了粒径为4~5 nm的纳米MgH2颗粒,在303 K温度下的吸氢量高达6.7%,MgH2的储氢热力学和动力学性能都得到大幅改善,放氢焓变为59.5 kJ/mol H2,吸/放氢活化能分别为28和80 kJ/mol H2。经过50 cyc循环后,仅损失0.07%的容量。另外,随着超声处理时间从4 h增加到12 h,尺寸分布峰逐渐从4~5 nm变化到15~16和20~50 nm,其放氢峰值温度依次分别为398、435和488 K,这证实了放氢特性与MgH2颗粒尺寸的依赖性。
一维和二维纳米结构镁基材料的储氢性能也有相关研究。Tan等[92]通过电子束蒸发技术在Al2O3和Si衬底上合成600 nm厚的Mg-4%Fe (原子分数)薄膜,这种Mg纳米薄膜的吸/放氢的焓值分别为(42.7 ± 3.2)和(66.9 ± 4.1) kJ/mol H2,远低于所制备的纳米颗粒的Mg/MgH2。Li等[21]利用气相沉积法制备直径为30~170 nm的Mg纳米线,发现30~50、80~100和150~170 nm的Mg纳米线解吸焓分别为65.3、65.9和67.2 kJ/mol H2,均小于块状MgH2。Li等[93]利用第一性原理计算研究发现减小Mg纳米线的尺寸会导致热力学不稳定,直径为0.85 nm的MgH2的解吸焓为34.54 kJ/mol H2,低于直径为1.24 nm的MgH2 (61.86 kJ/mol H2)。
纳米结构的镁基材料及其氢化物的吸/放氢动力学显著提高,主要是由于纳米尺寸导致氢扩散和解离途径缩短,以及表面/界面自由能的增加[21,75,76,92,94]。图4[21,75,76,94]为3种纳米结构的镁基材料(Mg纳米颗粒、Mg纳米薄膜和Mg纳米线)与325目Mg粉末(颗粒尺寸40~50 μm)的吸氢动力学。325目Mg粉末在573 K下30 min内吸氢量不足0.5%。化学还原法制备25 nm的Mg纳米颗粒60 s内可吸氢6%以上,且尺寸为32和38 nm的Mg颗粒分别在140和420 s内吸附最大容量的95%,吸氢速率随着颗粒尺寸的增大而降低。同样,纳米线与纳米薄膜结构相比微米Mg材料吸氢动力学也有显著的提升。
图4

图4 不同纳米结构Mg的吸氢动力学对比[21,75,76,94]
Fig.4 Comparisons of hydrogen absorption kinetics of different nanostructured Mg[21,75,76,94]
此外,研究者们对镁基储氢材料从纳米尺度进行设计,Mg/MgH2纳米颗粒被过渡金属、金属化合物或碳材料等包裹,制备了具有核/壳结构的纳米镁基储氢材料。其中,金属@碳复合核/壳结构的镁基复合材料对于提升材料的储氢热力学与动力学性能和循环稳定性效果最为显著[95]。Zhang等[96]在Mg颗粒中通过球磨工艺加入Ni负载石墨烯,石墨烯作为促进剂使Ni颗粒更均匀地分布在Mg的表面。Mg@Ni8Gn2在373 K下100 s内可吸氢6.28%,在523 K下30 min内放氢5.73%,且活化能降低到71.8 kJ/mol H2。Lotoskyy等[97]在Mg-Ti体系中添加石墨,石墨均匀地覆盖到MgH2表面,在100 cyc循环后吸/放氢速率和储氢量依然没有衰减。Jeon等[98]通过化学还原法合成了具有选择性气体渗透性的聚甲基丙烯酸甲酯(polymethyl methacrylate,PMMA)聚合物包覆纳米Mg颗粒的复合储氢材料,以高容量(约为Mg含量的6%,总含量的4%)实现在473 K下30 min内快速吸氢。上述核/壳结构纳米镁基复合材料储氢性能增强的原因在于:核/壳结构提供Mg与活性组分大量的接触界面和活性位点,提升H的传输速率;核壳结构界面处的原子存在相互作用,改变原子的电子结构,从而改善材料的热力学和动力学性能;同时核/壳结构可兼作保护剂,防止纳米镁基储氢材料的氧化。
3.1.4 非晶结构的优势
有序结晶MgH2中的Mg—H键强而稳定,氢占据位点有固定的类型和数量。相反,由于原子结构的无序,非晶相中间隙的大小和化学环境在一定范围内发生变化,会形成不同的氢占据位点,进而使Mg—H键不稳定。球磨制备的非晶态Mg2Ni-Ni复合材料在303 K、3 MPa下30 min内吸氢接近4%,实现了在室温开始吸氢,该复合材料非晶区域的氢化物的热力学稳定性低于结晶Mg2Ni合金,导致放氢温度大幅度降低[99]。非晶结构由于是完全无序结构,可作为氢渗透和扩散的途径,能够提高镁基材料吸/放氢动力学。非晶态Mg62Ni33Ca5合金在403 K、3.5 MPa下吸氢,在400 s以后,其吸氢速率高于结晶Mg62Ni33Ca5合金[100]。
事实上,大多数镁基非晶态储氢合金必须在高于其结晶温度的条件下进行放氢,因此在吸/放氢过程中无法避免结晶化。研究[101]发现,Mg11Y2Ni2非晶合金在573 K活化过程中结晶,晶化的Mg11Y2Ni2合金在523 K时5 min吸氢接近4.0%,10 min能放氢3.5%,依然保持较好的动力学。非晶态镁基合金经多次吸/放氢循环后,也会导致非晶相分离甚至结晶。如(Mg24Ni10Cu2)100 - x Nd x 非晶材料,在低于其晶化温度下进行10 cyc吸/放循环后观察到结晶Mg2Ni相,且氢化动力学也比初始非晶态时变慢[102]。因此,进一步研究氢诱导的非晶态结构变化及其对镁基非晶合金吸/放氢动力学性能的影响,对于理解镁基非晶态储氢合金的稳定性和获得良好的储氢性能十分重要。
Han等[103]将薄膜结构与非晶结构相结合,通过直流磁控溅射法制备3种厚度(300 nm、2 × 250 nm和10 × 50 nm,样品分别记为F300、F2*250和F10*50)的Mg85Ni14Ce1非晶合金薄膜,截面形貌分别如图5a~c[103]所示。所有薄膜样品的XRD谱中没有观察到尖锐的晶峰,说明均为非晶结构。3种厚度薄膜样品均能在393 K下实现放氢,F300、F2*250和F10*50氢化样品在10 h内放氢量分别达到1.2%、1.4%和0.85%,如图5d~f[103]所示。值得注意的是,该非晶薄膜结构在吸/放氢循环后表现出高稳定性。这表明,薄膜结构与纳米尺度相结合的非晶态镁基合金有望成为中等温度下可逆储氢应用的候选材料。
图5
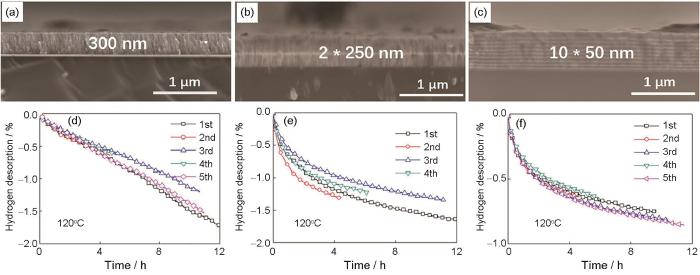
图5 Mg85Ni14Ce1非晶薄膜中F300、F2*250和F10*50样品截面的SEM像和393 K下的放氢动力学[103]
Fig.5 Cross-sectional SEM images (a-c) and kinetics of dehydrogenation at 393 K (d-f) of samples F300 (a, d), F2*250 (b, e), and F10*50 (c, f) in Mg85Ni14Ce1 amorphous films[103] (One Mg-Ni-Ce layer film in 300 nm, two Mg-Ni-Ce layers films in 250 nm, and ten Mg-Ni-Ce layers films in 50 nm were marked as F300, F2*250, and F10*50, respectively)
3.1.5 纳米玻璃
Xu等[104]报道了一种Mg-Ce-Ni-Cu纳米玻璃合金,该纳米玻璃合金是在熔体快淬制备非晶态合金的基础上进行HPT加工而成。从图6a[104]可以看出,熔体快淬Mg65Ce10Ni20Cu5合金的XRD谱是典型的非晶态,而HPT处理后样品含有少量的纳米晶相。图6b和c[104]表明,在393 K低温下,HPT处理1道次的合金可实现更快地吸氢,而熔体快淬样品在该温度下没有活性。这也表明HPT诱导的纳米玻璃形成(即纳米尺度上界面比例较大的非晶态相)提供了大量的氢扩散通道和活性位点,从而改善了吸氢动力学。
图6
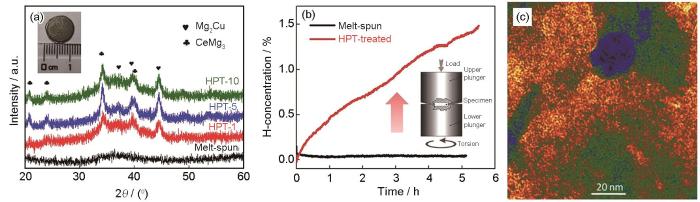
图6 Mg65Ce10Ni20Cu5合金经1、5和10道次HPT工艺前后的XRD谱,熔体快淬和HPT处理1道次后合金的吸氢动力学,及HPT处理1道次后样品的HRTEM像[104]
Fig.6 XRD spectra of melt-spun Mg65Ce10Ni20Cu5 alloy before and after HPT process for 1, 5, and 10 turns (inset: image of a disk after HPT process) (a), hydrogenation kinetics of sample processed by melt-spun and one HPT turns (b), and HRTEM image of sample processed by one HPT turns (c)[104] (Rotation speed ω = 0.5 r/min)
3.1.6 位错
位错作为晶体材料中内部的微观缺陷,能够有效改善镁基材料动力学性能。如1道次ECAP处理AZ31镁合金在材料中引入15.1 × 1015 m-2的高密度位错缺陷,比铸态样品表现出相对较快的吸氢动力学,吸氢速率可达0.0020%/s[71]。位错与H原子有很强的相互作用,由于刃型位错在金属中充当H原子陷阱的原因,在严重变形的金属中观察到氢溶解度的增强。有研究[105]表明高密度的刃型位错不仅可以作为氢扩散的途径,还可以增强氢化物相的形核和生长,利于储氢材料活化。
Edalati等[66]采用HPT处理纯Mg研究位错、晶界与吸氢动力学之间的联系。图7a~d[66]为退火及HPT处理转数N = 0.25和10时Mg样品的OM像和TEM像,Mg在N = 0.25时形成典型的剪切组织、亚晶粒及大量的位错;在N = 10时,微观组织为微/纳米尺寸的晶粒,晶粒中几乎无位错。通过吸氢动力学测试发现(图7e[66]),当N = 0.25时,吸氢量较小,与退火试样几乎没有显著差异。N = 10时,吸氢速率明显提高,比退火样品和N = 0.25 HPT处理样品快12倍,吸氢量达6.9%,吸氢速率提高归因于晶粒细化和高角度晶界分数的增加。这也说明SPD工艺在镁基材料中引入的位错缺陷,对于吸氢动力学性能提升的作用不如晶界显著。
图7

图7 HPT处理不同转数下Mg样品的OM像、TEM像和SAED花样,及退火样品和经HPT处理0.25和10转数样品的吸氢动力学曲线[66]
Fig.7 OM image for annealed Mg sample (a), TEM images and SAED patterns (insets) for Mg samples processed by HPT for 0.25 (b) and 10 (c, d) turns (Fig.7d is a dark-field image of Fig.7c), and hydrogen absorption kinetics curves of annealed samples and HPT treated samples at 0.25 and 10 turns (e)[66] (P—pressure, T—temperature, ε—equivalent strain)
3.1.7 层错
Hongo等[55]利用HPT + 退火处理的工艺方法处理Mg2Ni金属间化合物,在材料中引入层错(SF),如图8a~d[55]所示。条状线对应Mg2Ni的(001)面堆垛层错,TEM像模拟表明,亮线对应B层,暗线对应A层和C层。图8c和d[55]分别显示理想的ABCABC堆叠与一个代表性的堆垛层错缺陷。具有大量堆垛层错缺陷的样品在几分钟内吸氢超过2%,而没有堆垛层错缺陷的样品则表现出相对缓慢的氢化动力学(图8e[55])。堆垛层错与晶界作用类似,可作为将H从样品表面输送到内部的路径,图8f[55]为层错缺陷作为氢传输途径的示意图。另外,Mg-Ni-RE合金中的LPSO相在结构上实质也属于堆垛层错。引入堆垛层错的结构缺陷能改善镁基材料氢化动力学和活化,H在堆垛层错中的低扩散能垒,有助于H在镁基材料内部的扩散。
图8

图8 HPT处理10道次并在673 K退火后的Mg2Ni样品TEM像和HRTEM分析,有无堆垛层错缺陷的粗晶Mg2Ni的吸氢动力学曲线,及层错缺陷作为氢传输途径的示意图[55]
Fig.8 TEM images, hydrogen absorption kinetic curves, and schematic of Mg2Ni samples treated with 10 turns of HPT and annealed at 673 K[55]
(a) bright field image
(b) HRTEM image of lattice and corresponding SAED pattern (inset)
(c) normal ABCABC stacked lattice image
(d) lattice image with a stacking layer dislocation
(e) hydrogen absorption kinetic curves of coarse crystal Mg2Ni with and without stacking faults (t—annealed time)
(f) schematic of stacking faults as hydrogen transport pathways
3.1.8 择优取向
Botta等[52]通过HPT、CR、MS-CR工艺处理纯Mg,将其微观组织与吸/放氢性能相关联。尽管HPT晶粒细化效果最好,但HPT样品呈现出较弱(0001)织构,而CR和MS-CR样品中(0001)织构强度更强(图9a~c[52])。如图9d和e[52]所示,MS-CR和CR样品吸氢动力学远优于HPT样品,且2种样品均具有较好的放氢动力学,这与冷轧过程中产生的强(0001)织构有关。通过ECAP处理AZ31合金4道次后α-Mg相在不同的截面有明显的择优取向,横截面择优取向为(
图9

图9 不同工艺处理纯Mg的EBSD图和相应的极图,不同工艺下纯Mg活化后的吸氢动力学,及熔体快淬(MS)和熔体快淬-冷轧(MS-CR)样品的放氢动力学[52]
Fig.9 EBSD maps and corresponding polar maps of pure Mg treated by the processes of HPT (a), cold rolling (CR) (b), and MS-CR (c), kinetics of hydrogen absorption after Mg activation by different SPD processes (d), and kinetics of hydrogen desorption by MS and MS-CR samples (e)[52] (MS—melt spun, SPD—severe plastic deformation, RD—rolling direction, TD—transverse direction)
有研究[73]对(0001)织构提升镁基材料活化和动力学性能的机理给出解释:① 在(0001)取向大量存在的情况下,会优先在(0001)取向上形成氧化层,比(
3.2 表/界面对储氢性能的影响
3.2.1 表/界面
镁基材料的吸氢反应始于其表/界面,表/界面结构的特性决定着H2吸附和解离能力。Ham等[106]通过直流磁控溅射在刚性SiO2基底上合成Mg薄膜,随着薄膜厚度的增加,其形貌从连续的致密形貌转变为多孔柱状结构,Mg薄膜氢化物放氢的起始与峰值温度也逐渐升高。Mooij等[107]提出界面能会导致MgH2的不稳定,镁基材料的薄膜结构会引入额外的表/界面自由能,使系统自由能上升,理论计算和实验研究均表明表/界面能的引入能降低氢化物的放氢焓变。假设薄膜界面的厚度为1 nm,由此计算得到的额外界面自由能约为5 kJ/mol,再引入上述薄膜结构储存的额外自由能,Mg2NiH4和MgH2的生成焓分别下降到-59.5和-69.5 kJ/mol H2[108]。
除此之外,其他表面特性也会对镁基材料的储氢性能产生积极影响。Fujii等[109]用“协同现象”解释Mg/Pd多层膜低温吸/放氢的机理,Pd (催化剂)中H2的吸收会导致Mg膜中的拉应力,促进Mg膜的氢化;相反H2从Pd中放出导致压应力,使H2从MgH2中快速脱离。如“协同现象”所述,界面应力确实对氢吸附起着关键作用,可以实现Mg膜氢化物在373 K温度下放氢。Mg薄膜的氢吸附性能还与其表面晶体学特征相关。Ouyang等[110]通过控制溅射条件,分别合成具有(0001)取向、(0001)与(
采用喷涂技术制备具有催化作用的涂层修饰镁基材料表面,实现储氢性能的提高也有相关报道。El-Eskandarany等[111]采用冷喷涂工艺以Ni粉末涂覆冷轧Mg带材。图10a~f[111]为5.28%Ni粉末喷涂Mg带材的侧面和表面形貌及元素分布。Ni粉末在Mg带材表面发生塑性变形,形成多孔涂层。对比其他样品,3次喷涂冷轧300道次的Mg带材样品在423 K下5.1 min吸氢6.1%,473 K下21.1 min放氢6.1%,表现出优异的吸/放氢动力学,如图10g和h[111]所示。同时,该样品在473 K温度下经过600 cyc循环仍不会降低储氢容量,具有较好的长循环寿命特性。Ni粉末以超音速喷涂Mg带材的表面及亚表面,进一步地塑性变形产生孔隙与位错缺陷且实现了Mg和催化剂Ni之间的良好结合,提高了Mg带材吸/放氢动力学。
图10

图10 5.28%Ni (质量分数)粉末冷喷涂Mg带材后的侧面和表面的形貌及元素分布,423 K、8 × 105 Pa下不同样品的吸氢动力学,及473 K、2 × 104 Pa下不同样品的放氢动力学[111]
Fig.10 Morphologies and element distributions of the side (a-c) and surface (d-f) of 5.28%Ni (mass fraction) powder after cold spraying Mg strip, hydrogen absorption kinetics of different samples at 423 K, 8 × 105 Pa (g), and hydrogen desorption kinetics of different samples at 473 K, 2 × 104 Pa (h)[111] (SEI—secondary electron image)
3.2.2 微观界面
镁基材料内部的微观界面与H原子具有重要的相互作用。Karst等[112]基于MgH2特有的声子共振特性跟踪氢化物的形成、成核和生长,通过红外散射扫描近场光学显微技术(s-SNOM)研究原位环境下Mg/Ti/Pd-Au薄膜上Mg到MgH2的相变动力学。结果表明含大量晶界的Mg薄膜具有较快的吸氢动力学,氢化过程需要在晶界成核,晶界密度的增加加速了整个氢化过程,这可以解释纳米晶镁基材料体系中氢扩散系数的显著提高。事实上,不同取向大角度晶界(> 15°)由大量的位错和空位组成,晶界能量高于晶粒内部,有利于加速氢的扩散。另外,具有纳米晶的镁基材料也表现出优异的循环稳定性。ECAP处理4道次的ZK60镁合金,其晶粒尺寸细化至250 nm,在1000 cyc循环测试中没有表现出储氢容量或动力学方面的恶化,经实验研究及理论分析,这种稳定性主要归因于高密度的晶界与晶格缺陷为Mg/MgH2形核提供位点,而高密度的氢化物核又阻碍彼此的生长,保证了其结构和晶粒尺寸的稳定性[113]。
Fu等[57]通过超高压非平衡凝固制备具有低角度取向枝晶界面的Mg-5%Ni合金,证实了枝晶界面能够增强镁基材料的吸/放氢动力学。尽管形成的亚稳相Mg6Ni延迟其活化过程,仍然在枝晶界面中的{
相界在吸/放氢过程中也扮演着氢扩散途径的角色,如储氢相之间、储氢相与非储氢相之间以及储氢相-催化相之间界面。Song等[53]利用Mg-Ni-Y相图优化设计了具有多相共晶组织的Mg76.87Ni12.78Y10.35合金,由LPSO、Mg2Ni、MgNi4Y和Mg相组成。与铸造Mg2Ni合金相比,超细LPSO相和多相组织的形成提高了相界的体积分数,使得三元合金在活化和随后的吸氢动力学方面表现出显著的改善,Mg76.87Ni12.78Y10.35合金在573 K、3 MPa氢压首次活化1 min能吸氢3.0%[45]。另外,Mg与异相储氢材料之间的界面对氢的扩散和氢化物相形核也起到促进作用。累积叠轧(ARB)制备的Mg-LaNi5-Soot复合储氢材料具有良好的储氢特性,在573 K、0.2 MPa的平台压力下可逆储氢量达到4.5%,在573 K、2 MPa下30 s内可吸氢4.0%,快于ARB-Mg和Mg-LaNi5样品[114]。利用TEM对Mg-LaNi5-Soot复合材料的横截面微观结构及界面特征进行分析[115],图11a~d[115]为叠轧样品中储氢相Mg与LaNi5之间的界面关系,Mg与Mg界面呈波浪形,层间距在50~100 nm (图11a[115]);图11d[115]的衍射斑点证实分布于Mg晶界处的深色第二相颗粒为LaNi5相。图11e和f[115]为Mg与催化相Soot之间的界面关系,催化相颗粒尺寸约为100 nm,不仅存在于Mg-Mg界面处,且均匀地嵌入Mg基体中。图11g[115]为叠轧30道次样品和氢化30 s后样品横截面示意图,在2个Mg相之间的界面处存在LaNi5相与Soot颗粒,在氢化30 s后,LaNi5相为MgH2形核提供非均质成核位点。
图11
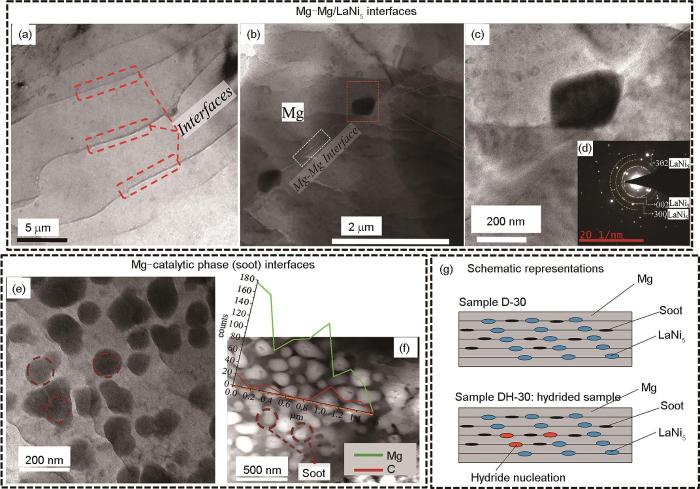
图11 累积叠轧(ARB) 30道次Mg-LaNi5-Soot样品TEM明场像(横截面),累积叠轧5道次Mg-LaNi5-Soot样品横截面TEM明场像,累积叠轧30道次Mg-Soot样品横截面TEM明场像和EDS线扫描叠加的暗场像,及ARB和氢化30 s后Mg-LaNi5-Soot样品横截面微观示意图[115]
Fig.11 TEM images and schematic representations of the cross-section of Mg-LaNi5-Soot obtained by accumulative roll bonding (ARB)[115]
(a) TEM bright-field image of Mg-LaNi5-Soot obtained by ARB 30 passes (cross-sectional view)
(b) TEM bright-field image of Mg-LaNi5-Soot obtained by ARB 5 passes
(c) zoomed-in view encompassing a single particle at the Mg-Mg interface
(d) SEAD pattern confirming the presence of LaNi5
(e) bright-field TEM image of Mg-Soot obtained by ARB 30 passes
(f) dark-field image with EDS line scan overlaid
(g) schematic representations of the cross-section of Mg-LaNi5-Soot samples
催化相与镁基储氢相之间形成的界面对材料的储氢性能同样有着重要的影响。Patelli等[116]利用He/H2气氛下将Mg和Ti蒸气相冷凝,在纳米尺度上形成了MgH2和TiH2相混合的双相纳米粒子。TiH2在吸/放氢过程中起到促进H2解离/重组和加速H原子扩散的催化作用,而Mg/MgH2提供H2的可逆储存。纳米颗粒中大量的Mg/TiH2界面形成了氢扩散的加速路径,提高Mg/MgH2吸/放氢动力学,在423 K时氢的吸附和解吸分别在不到100和1000 s内完成。此外,还证实了Mg/MgH2与TiH2界面区域的自由能是MgH2-TiH2双相纳米颗粒热力学稳定性降低的主要原因。Zhang等[117]在MgH2 + 9%Co@NiO复合材料中也观察到储氢相与催化相的活性界面起到催化与氢运输途径的作用,共生Mg2NiH4/Mg2CoH5团簇是由双金属催化剂前驱体原位形成并镶嵌在MgH2表面,被认为是“多级氢泵”,为氢吸收提供了表面途径。同时,MgH2与催化剂、Mg2NiH4/Mg2CoH5之间以及Mg与Mg2Ni/Mg2Co之间的界面为氢的扩散提供了大量的低能势垒通道,加速复合材料的吸/放氢。Aymonier等[118]利用超临界流体化学沉积(SFCD)工艺将纳米颗粒Ni沉积在Mg表面,研究纳米颗粒Ni催化剂与Mg之间的界面对储氢性能的影响。与球磨工艺中将Mg和Ni简单机械混合不同,在Mg与纳米Ni界面处形成了Mg2Ni界面相。正是这种界面相的存在使得SFCD制备得到的镁基复合材料吸氢动力学与可循环性要显著高于球磨工艺,在100 cyc循环后储氢量与动力学均未衰减。
4 结论与展望
镁基储氢相作为极具应用前景的储放氢介质,其种类、组织结构、界面等因素对储氢性能的影响至关重要。为调控储氢相及界面结构进而实现改善材料的吸/放氢热力学与动力学性能的目标,目前已经采用了多种制备工艺来加工与合成镁基储氢材料,包括熔体快淬、粉末冶金、球磨、大塑性变形、机械合金化、化学还原和气相沉积等。基于不同的制备工艺,本文总结出储氢相及其界面的调控效果对储氢热力学与动力学性能的作用规律:(1) 通过构筑二元或三元储氢相,能降低镁基材料的氢化物稳定性并提高吸/放氢动力学与循环稳定性;HPT工艺与热处理相结合,从不混溶材料体系中合成镁基亚稳相能够实现室温下可逆储放氢;具有亚稳结构(纳米/非晶结构)的储氢相由于纳米尺寸效应与非晶中原子结构的无序性也表现出更快的动力学和更优的热力学。(2) 大塑性变形对镁基储氢相组织和结构缺陷的适当调控设计,如引入择优取向、位错、层错和空位等,能够实现更好的活化、提高抗氧化性、快速的吸/放氢动力学和良好的循环稳定性,这也是经济上可行且环境上可接受的解决方案。(3) 储氢相表/界面结构的调控与修饰,如表面结构(储氢相表面形貌、取向和表面能等)和内部微观界面(晶界、相界及界面相等),为氢的捕获和扩散引入更多活性位点与迁移路径,利于提高镁基材料吸/放氢动力学与可循环性。(4) 制备亚稳结构的镁基储氢材料是开发其在温和条件下(温度373~473 K,氢压≤ 2 MPa)良好储氢性能的有效策略,但亚稳结构的稳定性使得材料的储氢可逆性存在一定的局限性,如镁基非晶合金在473 K以上及多次吸/放氢循环中会晶化,镁基亚稳相在较高温度会分解,纳米结构储氢相多次循环后会发生晶体生长与团聚,如何保证亚稳结构稳定性将直接影响镁基材料储氢热力学与动力学性能及循环寿命。因此,研究镁基材料中储氢相及其界面与储氢性能的相关性,未来仍然是开发高性能储氢材料的重要课题。
基于此,后续工作中仍需进一步解决的关键问题总结如下:(1) 目前已经有了大量的实验工作来探究不同镁基材料体系中的储氢相及表/界面结构与储氢性能之间的关系,但相关的理论模型和高通量预测依然比较少。通过将第一性原理计算、分子动力学模拟、CALPHAD等与大规模高通量筛选方法相结合,能够极大地减小新型镁基储氢材料的开发成本,并且可为高性能镁基储氢材料的设计和合成提供理论指导。(2) 在微合金化的基础上,通过设计化学成分、晶体结构、晶格缺陷等方式来调控镁基储氢相的亚稳态特性,实现储氢热力学和动力学性能的双重改性以及优异的循环稳定性;后续研究可侧重关注镁基储氢材料吸/放氢过程中界面结构的演化,如Mg-MgH2界面、镁基储氢相与非储氢相、异质储氢相及催化剂等之间的界面,对于深入理解表/界面结构与H2分子/H原子的交互作用机制具有重要意义。(3) 开发和优化方法简单、成本低廉的镁基储氢材料制备技术,在制备过程中引入额外的能量(如涉及到大塑性变形工艺的高剪切应变、高压和外部能量场等),使合成的新型结构镁基储氢材料(如纳米玻璃、纳米尺寸厚度的非晶薄膜和亚稳相等)吸/放氢热力学与动力学性能能够适应温和温度条件下(373~473 K) H2的储放;另外,用于表面处理与防护的涂层技术也可以为块体镁基储氢材料规模化生产开辟新的途径。(4) 新材料的开发和新工艺的提出,需要深入理解跨空间尺度和时间尺度的由组织缺陷主导的材料结构-储氢性能内在关系;借助基础的理论预测结合实验证实,筛选出对储氢性能提升起积极作用的关键储氢相及微观组织、表/界面结构;最终从目标储氢性能出发,以镁基储氢相及界面优化设计为准则,逆向设计具有容量高、吸/放氢速率快和寿命长的高性能镁基储氢材料。(5) 基于氢结合能工程,利用大塑性变形工艺设计与合成新的三元或三元以上镁基亚稳相,使其放氢温度降至中等温度甚至是室温。这种策略可以更普遍地应用于设计超越相平衡范围的具有优越储氢性能的新型镁基储氢材料。
来源--金属学报 沪公网安备31011202020290号
沪公网安备31011202020290号
