



分享:Fe-C-Mn三元合金中奥氏体-铁素体相变的相场模拟

张军
摘要
采用相场法研究了Fe-C-Mn三元合金在临界区等温过程中发生的奥氏体-铁素体相变。基于Gibbs自由能守恒理论,在相场模型中考虑了替换型元素Mn在奥氏体/铁素体相界面内扩散所导致的自由能耗散,描述了Fe-C-Mn三元合金中因Mn在相界面偏聚所导致的相变停滞和相变不完全现象。利用相场模型研究了Mn含量对奥氏体-铁素体相变微观组织转变和相变动力学的影响。结果表明,随着Mn含量的增加,铁素体相的转变速率和体积分数降低,Mn在奥氏体/铁素体相界面内扩散所导致的溶质拖曳现象越明显,相变不完全程度加剧。
关键词:
Fe-C合金中添加合金元素,如Mn、Ni、Cr等,有助于改善材料力学性能,对满足钢铁材料日益迫切的发展需求具有重要意义。奥氏体-铁素体相变是控制钢中相成分、晶粒尺寸及组织形貌等的重要转变之一。然而,在此相变过程中,由于合金元素的扩散速率远小于C元素,关于合金元素分配与否的问题,目前学术界仍存在争议[1~4]。近十年来,许多数学或物理模型描述了其相变动力学和元素分配行为。最早,Christian[2]提出的界面控制生长理论认为,相变驱动力完全用于界面迁移过程,由此,界面迁移速率的大小正比于相变驱动力和界面迁移率。而Zener[4]则认为相界面迁移所需要消耗的自由能可以忽略不计,相变过程完全由元素的扩散所控制。当然,Zener提出的扩散控制生长概念在二元合金的研究上得到了很好的证明。但是,对于三元Fe-C-X (X=Mn、Ni、Cr
Gamsjager[11]采用调整有效界面迁移率的方法来考虑X元素分配的影响,研究了Fe-C-X合金连续冷却过程中奥氏体-铁素体的混合控制相变过程,但其中有效界面迁移率与相变温度和冷却速率的关系很难确定。目前有2种理论:溶质拖曳[12]和自由能耗散理论[13],从热力学角度解释了X元素分配对相变动力学的影响。Hillert等[14,15]认为2种假设是等价的,即溶质原子在界面处的偏聚对移动界面产生的拖曳力等同于界面处溶质分配所耗散的自由能。基于上述假设,学者们建立了一些数学物理模型[16~18]来研究X元素对相变过程的影响,并通过合理假设描述了奥氏体-铁素体相变过程。然而,这些模型与相场法的结合却鲜有报道。相场法在介观尺度方面的研究又具有一些独特的优势, 例如,Loginova等[19]通过耦合溶质拖曳力模型,采用相场法研究了Fe-C合金中奥氏体-铁素体块状转变过程。Zhu等[20]运用相场法并结合自由能耗散模型,模拟了Fe-C-Mn合金中奥氏体-铁素体的循环相变过程。
相场法通过计算系列场变量描述多晶组织的微观结构和成分的演化。目前开发的相场模型已能成功地研究复杂微观几何结构和界面各向异性特征[21~23]对相变动力学和组织形貌的影响。同时,也能综合考虑有限C扩散与有限界面迁移率对相变动力学的影响[24~26],并成功地应用于奥氏体-铁素体混合控制相变过程的研究。而且,相场法能自动追踪界面位置的变化,也能更容易在界面处耦合溶质再分配的自由能耗散模型,分析界面处合金元素带来的影响。本工作采用多相场相变模型模拟Fe-C-Mn三元合金在临界区等温过程中发生的奥氏体-铁素体相变。通过2种相变模式,即考虑Mn分配和不考虑Mn分配,模拟单个铁素体晶粒的生长过程,分析Mn元素对相变动力学和转变体积分数的影响。进一步讨论合金中Mn含量对微观组织、C浓度以及相变动力学的影响。最后,针对铁素体相变不完全转变情况,分析不同Mn含量对铁素体相转变过程的影响机制。
在奥氏体-铁素体相变过程中,相较于C元素的扩散速率,合金元素Mn的扩散迁移速率非常缓慢。Mn元素在奥氏体相和铁素体相内的扩散速率也相差2~3量级, 故两相之间存在Mn的化学势阱,容易导致Mn元素在相界面处分配,对相界面的迁移起到拖曳作用,造成部分化学自由能被耗散。因此,Fe-C-Mn三元合金中相界面迁移驱动力的计算,需要考虑引入Mn元素分配的影响。
根据Gibbs自由能守恒理论[18],相变的化学驱动力需与相变过程中所消耗的能量相等。奥氏体-铁素体相变所消耗的化学自由能ΔGchem分为相界面迁移消耗的自由能ΔGfriction和耗散自由能ΔGdis 2部分:
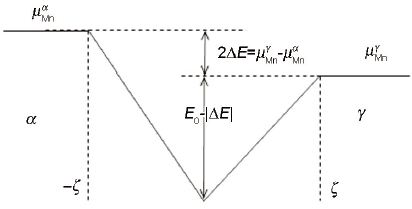
上式表明相界面迁移过程中共有ΔGdis的化学自由能不能作为相界面迁移的驱动力。Purdy等[12]假设Mn在奥氏体-铁素体相界面处存在楔形的作用势,如图1所示。界面处Mn元素在相界面的分配引起的ΔGdis可表述为:
式中,y为坐标位置,
式中,R为气体常数,T为相变温度,
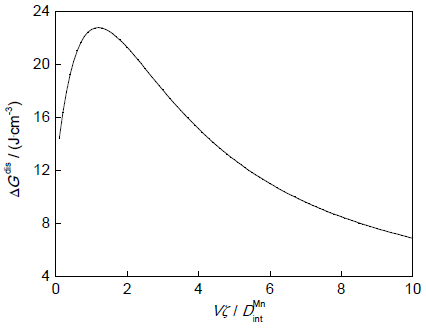
式中,Vm为奥氏体的摩尔体积。图2为E0=10 kJ/mol时Fe-0.1C-0.5Mn (质量分数,%)合金在T=1023 K时γ→α相变的ΔGdis与V的关系。可以发现:当高界面迁移速率时(>1 μm/s),ΔGdis较小,而在低界面迁移速率下作用明显。界面处自由能耗散会影响相变的动力学过程。
图1 相界面处Mn化学势的分布示意图
Fig.1 Schematic of the chemical potential of Mn inside the austenite/ferrite interface (2ζ is the physical interface thickness, E0 is the Mn interface binding energy,
图2 γ→α相变过程中耗散自由能(ΔGdis)的变化
Fig.2 Gibbs-energy dissipation (ΔGdis) as a function of
本工作采用多相场模型模拟Fe-C-Mn三元合金的奥氏体-铁素体的相变过程,模型中选取非保守场变量
体系内的系统自由能F为序参量
式中,Fs和Fc分别为界面自由能和化学自由能,两者均为相场变量
为了更直观地描述界面处Mn分配带来的自由能耗散对相变的影响,本工作采用2种相变模式对奥氏体-铁素体相变过程进行了模拟。模式I:相场模型仅考虑体系内C原子的长程扩散行为,不考虑自由能耗散;模式II:相场模型考虑体系内C原子的长程扩散行为和界面处Mn分配带来的自由能耗散。
C浓度场变量xC(r, t)的演化采用Cahn-Hilliard动力学方程[27,28]来描述:
序参量ηki(r, t)的演化采用Allen-Cahn动力学方程[27,28]来描述:
模式I:
模式II:
式中,δ为相场界面宽度;L和Mk分别为与界面迁移率Mp和C元素扩散迁移率
模拟中假设相变过程中相界面处相之间的C化学势平衡,C原子可在两相间快速交换:
采用规整溶体双亚点阵模型[24,29,30]计算Fe-C-Mn三元体系中奥氏体相与铁素体相的化学自由能,两相之间的化学自由能差ΔGchem提供奥氏体-铁素体相变的驱动力。
本工作针对Fe-C-Mn三元合金在临界区等温过程中发生的奥氏体-铁素体相变进行模拟。文中各元素所使用的浓度单位都为质量分数(%)。初始组织中奥氏体内的C和Mn均匀分布,而铁素体晶核的初始C浓度设为0.01%。采用有限差分和逐步迭代方法对动力学方程进行求解。为提高数值计算的稳定性及精确性,在离散相场变量η和xC时,考虑了第一和二近邻的影响。V的计算通过对序参量η在时间(Δt)和空间(Δy)上进行一阶离散来求解:
其中,Δy为网格单元尺寸,Δt为时间步长。为满足计算精度要求,对模拟区域进行离散时,需预设的相场界面宽度δ与Δy的关系为[27]:δ=nΔy (其中,n为取向数)。当4≤n≤6时,计算误差可控制在5.0%以内,本模拟设定Δy=0.4 μm,n=6。模拟所采用的物理参数如表1所示。
表1 模拟所采用的物理参数
Table 1 Physical parameters used in the simulations
3.1 2种相变模式下的相变行为
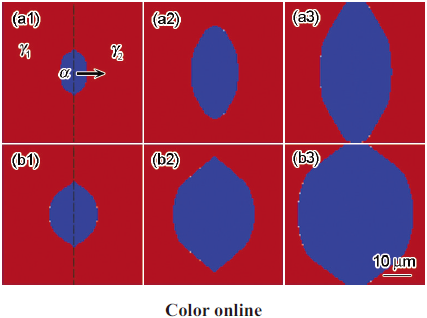
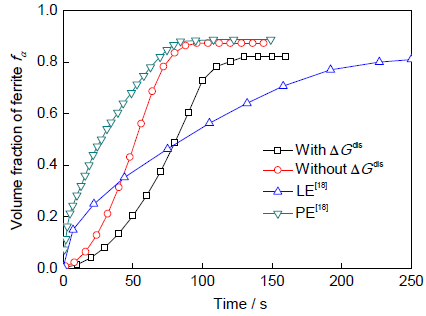
模拟所用材料选为Fe-0.1C-1.0Mn合金。图3为973 K等温时单个铁素体晶粒在2个奥氏体晶粒间生长的模拟结果。从图中可以发现,铁素体晶粒沿原奥氏体晶界比垂直于原奥氏体晶界方向生长快,呈椭圆状。比较图3a和b的模拟结果发现,图3a的结果中铁素体晶粒生长较慢,并且,铁素体晶粒沿奥氏体晶界与垂直于奥氏体晶界方向之间的生长速率差距较大。图4为铁素体相的转变动力学。在973 K下模拟结果与文献[18]报道结果比较可以发现,模式I模拟的相变体积分数与文献报道的PE模型的结果接近,而模式II模拟的体积分数与LE模型的结果接近。另外,比较模式I和II的铁素体转变动力学发现,2者在相转变速率和体积分数上表现出明显差异:后者的相转变速率明显慢于前者,而且,后者的铁素体相体积分数相对较小。2者不同之处仅在于是否引入ΔGdis的影响,即是否考虑Mn分配对界面处相变驱动力的影响。
图3 973 K等温时单个铁素体晶粒的生长过程
Fig.3 Single ferrite α growths between austenite grains γ1 and γ2 at 973 K with ΔGdis (a1~a3) and without ΔGdis (b1~b3) at time t =20 s (a1, b1), t =40 s (a2, b2) and t =60 s (a3, b3)
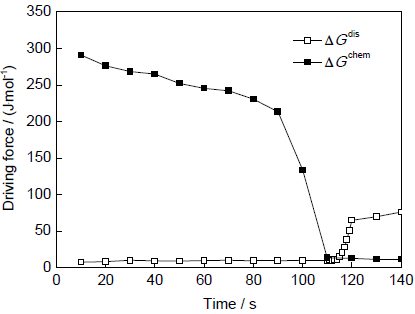
图5为相界面处消耗的化学自由能ΔGchem与耗散自由能ΔGdis随时间的演化情况。图5中的耗散自由能ΔGdis为图3a中铁素体沿箭头指示方向生长时相界面处的耗散自由能,即采用此生长方向的相界面迁移速率计算出的耗散自由能。可以看出,随着相变的进行,ΔGchem逐渐降低,此时相界面迁移速率相对较快,ΔGdis的作用并不明显。但随着界面迁移速率的降低,ΔGdis的作用越来越明显。当ΔGchem≤ΔGdis,即ΔGfriction≤0时,α/γ相界面会停止迁移,意味着相变过程发生停滞。但此时的ΔGchem仍大于零,说明并未达到热力学平衡状态,此时的相变停滞是因Mn元素在相界面分配所引起的,这种暂时的相变停滞会导致相变的不完全[18,20,33]。
图4 973 K等温时铁素体晶粒的生长动力学
Fig.4 Kinetics of ferrite α growth under different conditions at 973 K, comparing with the LE and PE predictions[
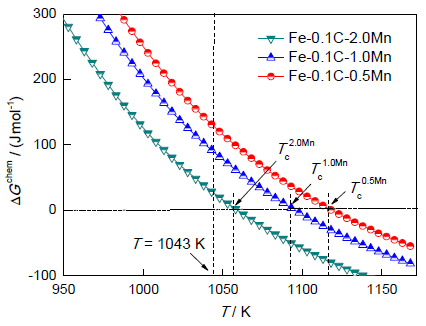
图6为3种合金Fe-0.1C-mMn (m=2.0、1.0和0.5)的奥氏体-铁素体相变的化学驱动力ΔGchem随相变温度T变化的情况。可见,相变驱动力随合金中Mn含量的增加而降低。另外,图6表明不同合金中奥氏体-铁素体相变发生的临界温度Tc随Mn含量的增加而降低,故选择的模拟温度必须在合金的临界温度之下,同时模拟温度不能过低,否则其相变驱动力过大,从而导致不易区分不同合金相变后的微观组织形貌。而且,在奥氏体-铁素体相变过程中,考虑铁素体形核仅发生在原始奥氏体晶界上,形核率的计算可参考文献[28]。
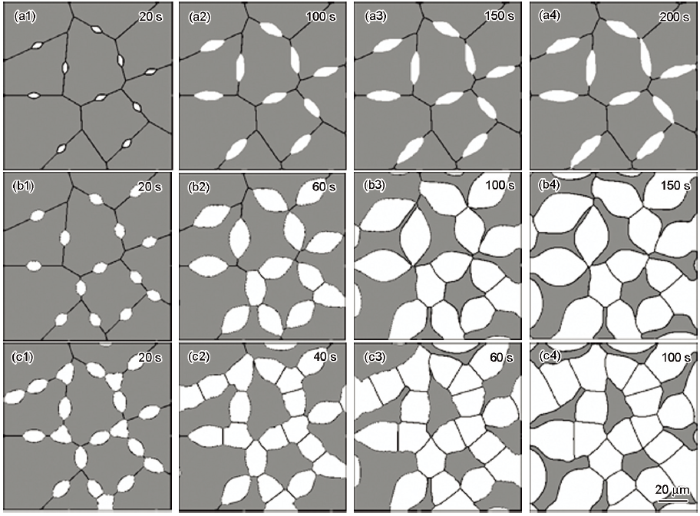
为研究Mn含量对奥氏体-铁素体相变过程的影响,采用模式II针对此3种合金在1043 K进行等温奥氏体-铁素体相变模拟,其微观组织演化的模拟结果如图7所示。图中灰色区域为原奥氏体相,白色区域代表新形成的铁素体相。可以发现:(1) 0.5Mn成分下铁素体晶粒生长迅速,而且沿原奥氏体晶界比垂直于原奥氏体晶界方向生长快;(2) 1.0Mn成分下铁素体晶粒生长速率降低,但沿原奥氏体晶界与垂直于原奥氏体晶界方向的生长速率之间差距增大;(3) 2.0Mn成分下铁素体晶粒生长最慢,但沿原奥氏体晶界的生长速率远大于垂直于原奥氏体晶界方向的生长速率。相变完成时,微观组织表现出明显的差异:在0.5Mn成分下,由于铁素体晶粒之间接触较早,铁素体晶粒沿各方向的距离并不相等,最终形成为不规则状铁素体;在1.0Mn成分下,铁素体晶粒接触减少,并沿各方向充分生长,最终形成为椭圆状铁素体;在2.0Mn成分下,铁素体晶粒之间接触很少,并沿着原奥氏体晶界快速生长,最终形成为细长铁素体。观察3种成分不同时刻的铁素体晶粒组织,可以看出,相变过程中铁素体晶粒生长形貌主要受到不同方向上生长速率变化的影响。生长速率的大小主要依赖于相界面迁移驱动力的大小。图6所示T=1043 K时Fe-0.1C-2.0Mn合金的化学驱动力约为30 J/mol,比Fe-0.1C-1.0Mn合金的要小约60 J/mol,2.0Mn成分下相界面迁移速率最低,铁素体晶粒生长最慢。并且,Mn元素分配对移动界面产生拖曳作用,消耗部分自由能ΔGdis,致使相界面迁移驱动力大幅降低,如此减弱了化学驱动力对铁素体晶粒生长的影响,而增强了界面能的影响, 这种效应随着合金中Mn含量的增大而逐渐增强,导致沿原奥氏体晶界与垂直于原奥氏体晶界方向的生长速率之间的差距逐渐增大,铁素体晶粒形貌也会从椭圆状向扁平状转变。
图5 相界面迁移过程中ΔGchem与ΔGdis的变化情况
Fig.5 Variations of total dissipation of the chemical free energy (ΔGchem) and ΔGdis with time due to Mn diffusion inside the interface for austenite-to-ferrite transformation at 973 K
图6 不同合金的奥氏体-铁素体化学驱动力(ΔGchem)与相变温度(T)的关系
Fig.6 Chemical driving forces for the austenite-to-ferrite transformation as a function of temperature (T) in the Fe-0.1C-2.0Mn, Fe-0.1C-1.0Mn and Fe-0.1C-0.5Mn (mass fraction, %) alloys (Tc—triggering temperature of γ→α transformation)
图7 1043 K等温时不同合金相变过程中微观组织演化的模拟结果
Fig.7 Simulations of temporal evolutions of the microstructures at 1043 K during the austenite-to-ferrite transformation for Fe-0.1C-2.0Mn (a1~a4) , Fe-0.1C-1.0Mn (b1~b4) and Fe-0.1C-0.5Mn (c1~c4)
图8为3种合金相变过程中C浓度场演化的模拟结果。为了清晰辨识不同合金成分下C浓度的分布情况,图中采用了不同C浓度标尺对模拟结果进行标示。从图8中可以看出,Mn含量越高,奥氏体内形成的C浓度越低。Fe-0.1C-2.0Mn、Fe-0.1C-1.0Mn和Fe-0.1C-0.5Mn合金的C浓度(%)分别为0.115、0.271和0.371,皆低于THERMOCALC热力学计算的结果0.177、0.301和0.386 (%)。另外,Mn含量的高低也将影响相变过程中奥氏体内C元素的分布均匀性。在Fe-C-Mn三元合金中两相区等温过程中形成组织的Mn和C成分的高低及分布,对后续的连续冷却过程中贝氏体或马氏体相变有重要影响。
图8 1043 K等温时不同合金相变过程中C浓度场演化的模拟结果
Fig.8 Simulations of temporal evolutions of the carbon concentration fields at 1043 K during the austenite-to-ferrite transformation for Fe-0.1C-2.0Mn (a1~a4) , Fe-0.1C-1.0Mn (b1~b4) and Fe-0.1C-0.5Mn (c1~c4)
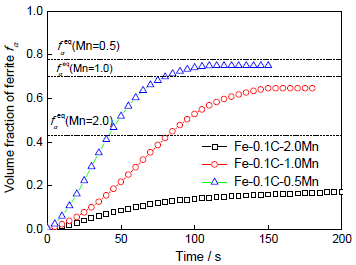
图9 不同合金在1043 K等温时的转变动力学
Fig.9 Ferrite fractions as a function of time during isothermal austenite-to-ferrite transformation at 1043 K for different alloys (
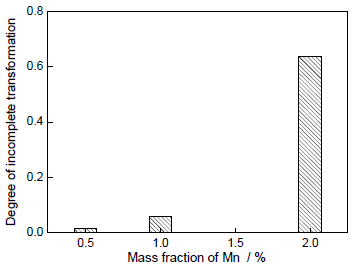
图10 不同Mn含量下相变不完全的转变程度
Fig.10 Degrees of incomplete transformation at 1043 K as a function of Mn concentration
图9所示为3种合金的铁素体相转变体积分数fα随时间的演化情况。可以看出,相变动力学受到合金中Mn含量的影响,Mn含量越低,相变进行越快,完成所需时间越短,如0.5Mn成分下相变仅需要100 s就完成,而在2.0Mn成分下在t=200 s时还未完成。另外,不同Mn含量下形成的铁素体体积分数不同:Mn含量越高,形成的铁素体体积分数越低, 但此时3种合金中的相变均未达到完全平衡。
定义
合金中Mn含量的增加,不仅导致相变驱动力ΔGchem的降低,而且造成界面处Mn分配过程中所耗散的自由能ΔGdis更大,两者的共同作用致使相界面迁移驱动力ΔGfriction降低,造成铁素体生长受阻碍程度增加,因此相变过程更早出现生长停滞,且相变不完全的转变程度加剧。所以,Fe-C-Mn三元合金的相变过程不仅受C原子的长程扩散行为所控制,而且也受到界面处Mn分配的影响。合金中Mn元素的添加不仅会影响奥氏体-铁素体相变过程中铁素体晶粒的生长形貌,而且会改变其相变动力学。
(1) 本相场模型引入了自由能耗散模型,模拟了Fe-C-Mn三元合金在混合控制生长模式下的奥氏体-铁素体相变过程。通过2种相变模式对比,发现该相变过程中界面处Mn元素的分配行为会影响相转变的热力学和动力学,导致相变出现停滞现象。
(2) 合金中的Mn含量会影响铁素体晶粒的生长形貌。Mn含量越高,铁素体晶粒沿原奥氏体晶界与垂直于原奥氏体晶界方向的生长速率之间的差距越大,晶粒形貌从椭圆状向扁平状转变。此外,随着Mn含量的升高,奥氏体相中C浓度与理论平衡C浓度之间的差距增大。
(3) 铁素体相的转变动力学和体积分数随合金中Mn含量的升高而降低。高Mn含量导致相变驱动力降低和Mn元素分配所耗散的自由能ΔGdis增大,使相变不完全的转变程度加剧。
 , 李殿中
, 李殿中
1 模型
1.1 自由能耗散模型
1.2 相场变量与相场方程
2 模拟条件
Parameter (unit)
Value
Ref.
0.4
[20,31]
0.79
[31]
[20]
[32]
[32]
[32]
3 模拟结果及分析
3.2 Mn元素含量的影响

4 结论
来源--金属学报
 沪公网安备31011202020290号
沪公网安备31011202020290号
